Dinesh Giri, Daniel Rigden, Mohammed Didi, Matthew Peak, Paul McNamara, Senthil Senniappan
{"title":"Novel compound heterozygous <i>ASXL3</i> mutation causing Bainbridge-ropers like syndrome and primary IGF1 deficiency.","authors":"Dinesh Giri, Daniel Rigden, Mohammed Didi, Matthew Peak, Paul McNamara, Senthil Senniappan","doi":"10.1186/s13633-017-0047-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>De novo truncating and splicing mutations in the additional sex combs-like 3 (<i>ASXL3</i>) gene have been implicated in the development of Bainbridge-Ropers syndrome (BRPS) characterised by severe developmental delay, feeding problems, short stature and characteristic facial features.</p><p><strong>Case presentation: </strong>We describe, for the first time, a patient with severe short stature, learning difficulties, feeding difficulties and dysmorphic features with a novel compound heterozygous mutation in <i>ASXL3</i>.Additionally the patient also has primary insulin like growth factor-1 (IGF1) deficiency. The mutations occur in exon 11 and proximal part of exon 12 and are strongly conserved at the protein level across various species. <i>In-silico</i> analyses using PolyPhen-2 and SIFT predict the amino acid substitutions to be potentially deleterious to the protein function. Detailed bioinformatics analysis show that the molecular defects caused by the two compound heterozygous mutations synergistically impact on two points of the molecular interaction network of <i>ASXL3.</i></p><p><strong>Conclusion: </strong>We hypothesise that <i>ASXL3</i> potentially has a role in transcriptional activation of <i>IGF1</i> involved in signalling pathways that regulate cell proliferation and growth, which could be contributing to short stature encountered in these patients.</p>","PeriodicalId":14271,"journal":{"name":"International Journal of Pediatric Endocrinology","volume":"2017 ","pages":"8"},"PeriodicalIF":0.0000,"publicationDate":"2017-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13633-017-0047-9","citationCount":"12","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13633-017-0047-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/8/4 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 12
Abstract
Background: De novo truncating and splicing mutations in the additional sex combs-like 3 (ASXL3) gene have been implicated in the development of Bainbridge-Ropers syndrome (BRPS) characterised by severe developmental delay, feeding problems, short stature and characteristic facial features.
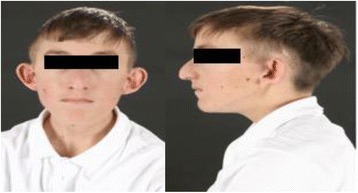
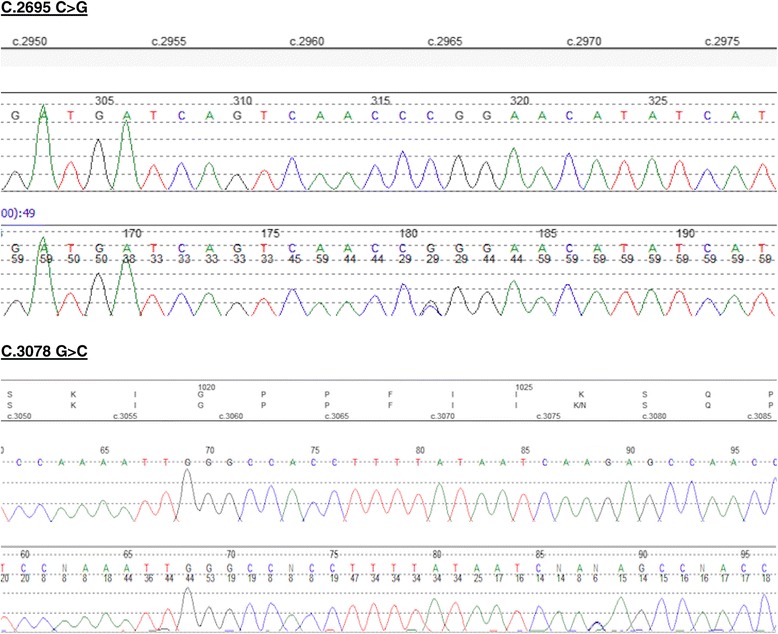
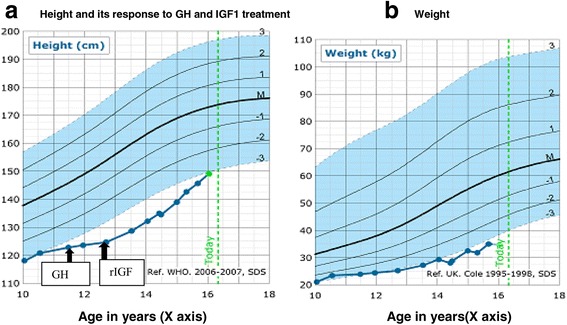
Case presentation: We describe, for the first time, a patient with severe short stature, learning difficulties, feeding difficulties and dysmorphic features with a novel compound heterozygous mutation in ASXL3.Additionally the patient also has primary insulin like growth factor-1 (IGF1) deficiency. The mutations occur in exon 11 and proximal part of exon 12 and are strongly conserved at the protein level across various species. In-silico analyses using PolyPhen-2 and SIFT predict the amino acid substitutions to be potentially deleterious to the protein function. Detailed bioinformatics analysis show that the molecular defects caused by the two compound heterozygous mutations synergistically impact on two points of the molecular interaction network of ASXL3.
Conclusion: We hypothesise that ASXL3 potentially has a role in transcriptional activation of IGF1 involved in signalling pathways that regulate cell proliferation and growth, which could be contributing to short stature encountered in these patients.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
