Salwa A Musa, Areej A Ibrahim, Samar S Hassan, Matthew B Johnson, Asmahan T Basheer, Ali M Arabi, Mohamed A Abdullah
{"title":"Fanconi Bickel syndrome: clinical phenotypes and genetics in a cohort of Sudanese children.","authors":"Salwa A Musa, Areej A Ibrahim, Samar S Hassan, Matthew B Johnson, Asmahan T Basheer, Ali M Arabi, Mohamed A Abdullah","doi":"10.1186/s13633-020-00091-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Fanconi-Bickel syndrome (FBS) is a rare condition of carbohydrate metabolism, caused by a recessive defect in the facilitative glucose transporter GLUT2 encoded by the SLC2A2 gene and characterized by a wide spectrum of phenotypical features. There is a paucity of reported data on FBS from Sub-Saharan Africa. Here, we describe the clinical, biochemical and genetic characteristics of our patients with FBS from Sudan, a country with a high consanguinity rate.</p><p><strong>Patients & methods: </strong>Eleven patients from ten unrelated Sudanese families were included. Clinical & biochemical data were documented and imaging studies done including bone survey and abdominal ultrasound. Liver biopsy was done to confirm the pathological diagnosis in 45% of cases and molecular genetics was performed through contribution with the Exeter genomics laboratory for ten patients.</p><p><strong>Results: </strong>Reported consanguinity was 70% among our patients. Growth was significantly impaired at presentation with mean weights of (-5.3 ± 1.8) SD and heights (-5.4 ± 2.5) SD. Severe chest deformity was present in (27%) and all patients showed features of rickets at presentation. Three patients had neonatal diabetes requiring insulin therapy of which one has been reported before. Six families lost undiagnosed siblings with similar clinical presentations. We identified a total of four homozygous pathogenic SLC2A2 variants in our patients, one of whom had a novel mutation.</p><p><strong>Conclusions: </strong>FBS is not uncommon in Sudan where there is a high rate of consanguinity. Many cases are likely missed because of variable presentation and lack of public and professionals' awareness. This is the first series to describe this condition from Sub-Saharan Africa.</p>","PeriodicalId":14271,"journal":{"name":"International Journal of Pediatric Endocrinology","volume":"2020 1","pages":"21"},"PeriodicalIF":0.0000,"publicationDate":"2020-11-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13633-020-00091-5","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13633-020-00091-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 4
Abstract
Background: Fanconi-Bickel syndrome (FBS) is a rare condition of carbohydrate metabolism, caused by a recessive defect in the facilitative glucose transporter GLUT2 encoded by the SLC2A2 gene and characterized by a wide spectrum of phenotypical features. There is a paucity of reported data on FBS from Sub-Saharan Africa. Here, we describe the clinical, biochemical and genetic characteristics of our patients with FBS from Sudan, a country with a high consanguinity rate.
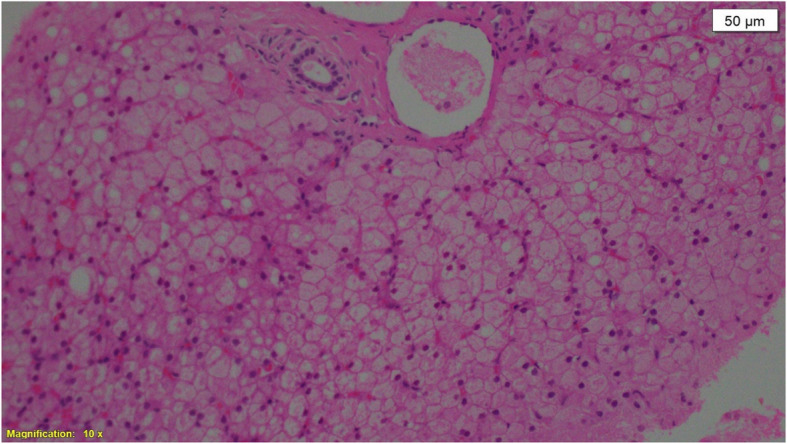
Patients & methods: Eleven patients from ten unrelated Sudanese families were included. Clinical & biochemical data were documented and imaging studies done including bone survey and abdominal ultrasound. Liver biopsy was done to confirm the pathological diagnosis in 45% of cases and molecular genetics was performed through contribution with the Exeter genomics laboratory for ten patients.
Results: Reported consanguinity was 70% among our patients. Growth was significantly impaired at presentation with mean weights of (-5.3 ± 1.8) SD and heights (-5.4 ± 2.5) SD. Severe chest deformity was present in (27%) and all patients showed features of rickets at presentation. Three patients had neonatal diabetes requiring insulin therapy of which one has been reported before. Six families lost undiagnosed siblings with similar clinical presentations. We identified a total of four homozygous pathogenic SLC2A2 variants in our patients, one of whom had a novel mutation.
Conclusions: FBS is not uncommon in Sudan where there is a high rate of consanguinity. Many cases are likely missed because of variable presentation and lack of public and professionals' awareness. This is the first series to describe this condition from Sub-Saharan Africa.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
