S. Stasik , C. Schuster , C. Ortlepp , U. Platzbecker , M. Bornhäuser , J. Schetelig , G. Ehninger , G. Folprecht , C. Thiede
{"title":"An optimized targeted Next-Generation Sequencing approach for sensitive detection of single nucleotide variants","authors":"S. Stasik , C. Schuster , C. Ortlepp , U. Platzbecker , M. Bornhäuser , J. Schetelig , G. Ehninger , G. Folprecht , C. Thiede","doi":"10.1016/j.bdq.2017.12.001","DOIUrl":null,"url":null,"abstract":"<div><p>Monitoring of minimal residual disease (MRD) has become an important clinical aspect for early relapse detection during follow-up care after cancer treatment. Still, the sensitive detection of single base pair point mutations via Next-Generation Sequencing (NGS) is hampered mainly due to high substitution error rates. We evaluated the use of NGS for the detection of low-level variants on an Ion Torrent PGM system. As a model case we used the c.1849G > T (p.Val617Phe) mutation of the <em>JAK2</em>-gene. Several reaction parameters (e.g. choice of DNA-polymerase) were evaluated and a comprehensive analysis of substitution errors was performed. Using optimized conditions, we reliably detected <em>JAK2</em> c.1849G > T VAFs in the range of 0.01–0.0015% which, in combination with results obtained from clinical data, validated the feasibility of NGS-based MRD detection. Particularly, PCR-induced transitions (mainly G > A and C > T) were the major source of error, which could be significantly reduced by the application of proofreading enzymes. The integration of NGS results for several common point mutations in various oncogenes (i.e. <em>IDH1</em> and <em>2</em>, c-<em>KIT</em>, <em>DNMT3A</em>, <em>NRAS</em>, <em>KRAS</em>, <em>BRAF</em>) revealed that the prevalent transition vs. transversion bias (3.57:1) has an impact on site-specific detection limits of low-level mutations. These results may help to select suitable markers for MRD detection and to identify individual cut-offs for detection and quantification.</p></div>","PeriodicalId":38073,"journal":{"name":"Biomolecular Detection and Quantification","volume":"15 ","pages":"Pages 6-12"},"PeriodicalIF":0.0000,"publicationDate":"2018-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.bdq.2017.12.001","citationCount":"34","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomolecular Detection and Quantification","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2214753517302115","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 34
Abstract
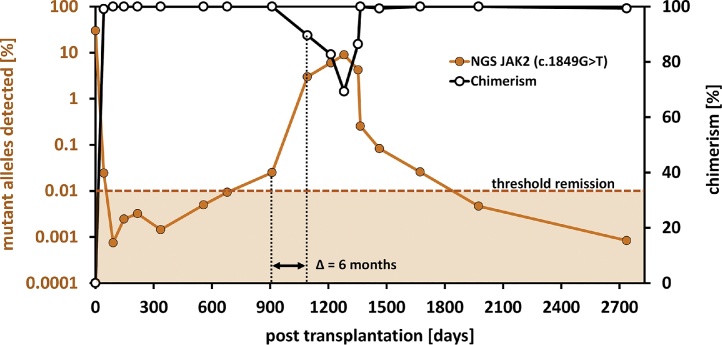
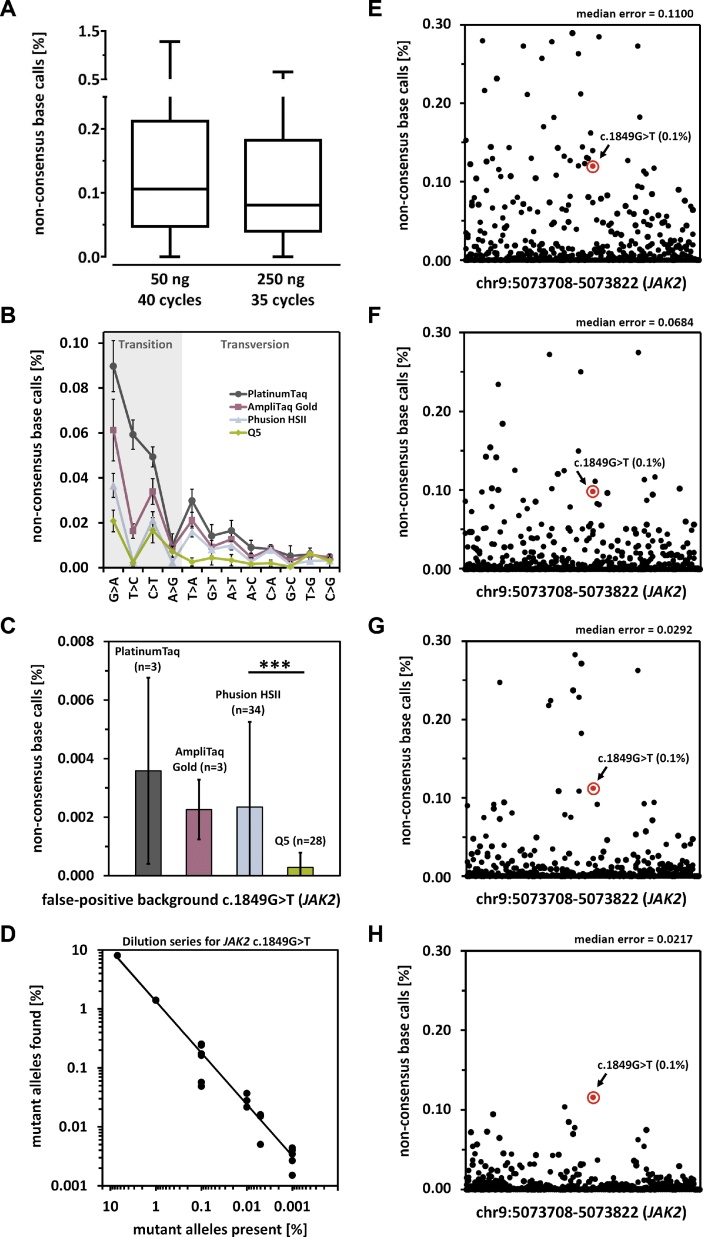
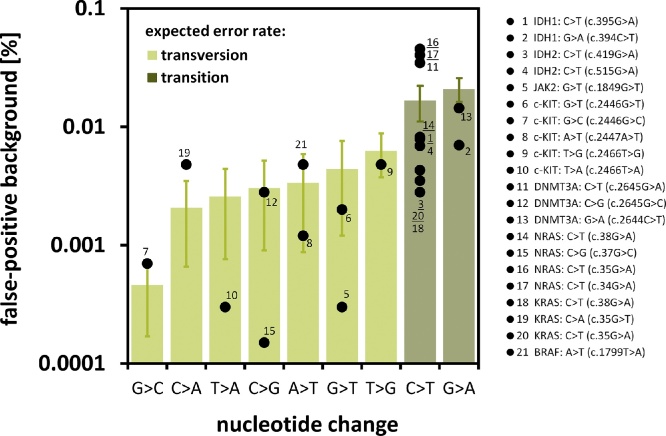
Monitoring of minimal residual disease (MRD) has become an important clinical aspect for early relapse detection during follow-up care after cancer treatment. Still, the sensitive detection of single base pair point mutations via Next-Generation Sequencing (NGS) is hampered mainly due to high substitution error rates. We evaluated the use of NGS for the detection of low-level variants on an Ion Torrent PGM system. As a model case we used the c.1849G > T (p.Val617Phe) mutation of the JAK2-gene. Several reaction parameters (e.g. choice of DNA-polymerase) were evaluated and a comprehensive analysis of substitution errors was performed. Using optimized conditions, we reliably detected JAK2 c.1849G > T VAFs in the range of 0.01–0.0015% which, in combination with results obtained from clinical data, validated the feasibility of NGS-based MRD detection. Particularly, PCR-induced transitions (mainly G > A and C > T) were the major source of error, which could be significantly reduced by the application of proofreading enzymes. The integration of NGS results for several common point mutations in various oncogenes (i.e. IDH1 and 2, c-KIT, DNMT3A, NRAS, KRAS, BRAF) revealed that the prevalent transition vs. transversion bias (3.57:1) has an impact on site-specific detection limits of low-level mutations. These results may help to select suitable markers for MRD detection and to identify individual cut-offs for detection and quantification.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
