{"title":"Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy.","authors":"John Hyun Namgoong, Carmen Bertoni","doi":"10.2147/DNND.S71808","DOIUrl":null,"url":null,"abstract":"<p><p>Duchenne muscular dystrophy (DMD) is an autosomal dominant, X-linked neuromuscular disorder caused by mutations in dystrophin, one of the largest genes known to date. Dystrophin gene mutations are generally transmitted from the mother to male offspring and can occur throughout the coding length of the gene. The majority of the methodologies aimed at treating the disorder have focused on restoring a shorter, although partially functional, dystrophin protein. The approach has the potential of converting a severe DMD phenotype into a milder form of the disease known as Becker muscular dystrophy. Others have focused on ameliorating the disease by targeting secondary pathologies such as inflammation or loss of regeneration. Of great potential is the development of strategies that are capable of restoring full-length dystrophin expression due to their ability to produce a normal, fully functional protein. Among these strategies, the use of read-through compounds (RTCs) that could be administered orally represents an ideal option. Gentamicin has been previously tested in clinical trials for DMD with limited or no success, and its use in the clinic has been dismissed due to issues of toxicity and lack of clear benefits to patients. More recently, new RTCs have been identified and tested in animal models for DMD. This review will focus on one of those RTCs known as ataluren that has now completed Phase III clinical studies for DMD and at providing an overview of the different stages that have led to its clinical development for the disease. The impact that this new drug may have on DMD and its future perspectives will also be described, with an emphasis on the importance of further assessing the clinical benefits of this molecule in patients as it becomes available on the market in different countries.</p>","PeriodicalId":11147,"journal":{"name":"Degenerative Neurological and Neuromuscular Disease","volume":"6 ","pages":"37-48"},"PeriodicalIF":0.0000,"publicationDate":"2016-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/DNND.S71808","citationCount":"17","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Degenerative Neurological and Neuromuscular Disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/DNND.S71808","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 17
Abstract
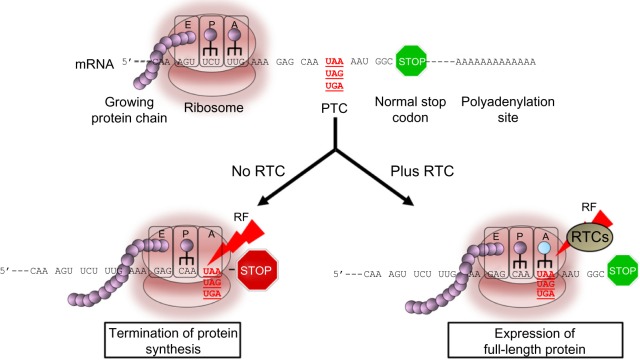
Duchenne muscular dystrophy (DMD) is an autosomal dominant, X-linked neuromuscular disorder caused by mutations in dystrophin, one of the largest genes known to date. Dystrophin gene mutations are generally transmitted from the mother to male offspring and can occur throughout the coding length of the gene. The majority of the methodologies aimed at treating the disorder have focused on restoring a shorter, although partially functional, dystrophin protein. The approach has the potential of converting a severe DMD phenotype into a milder form of the disease known as Becker muscular dystrophy. Others have focused on ameliorating the disease by targeting secondary pathologies such as inflammation or loss of regeneration. Of great potential is the development of strategies that are capable of restoring full-length dystrophin expression due to their ability to produce a normal, fully functional protein. Among these strategies, the use of read-through compounds (RTCs) that could be administered orally represents an ideal option. Gentamicin has been previously tested in clinical trials for DMD with limited or no success, and its use in the clinic has been dismissed due to issues of toxicity and lack of clear benefits to patients. More recently, new RTCs have been identified and tested in animal models for DMD. This review will focus on one of those RTCs known as ataluren that has now completed Phase III clinical studies for DMD and at providing an overview of the different stages that have led to its clinical development for the disease. The impact that this new drug may have on DMD and its future perspectives will also be described, with an emphasis on the importance of further assessing the clinical benefits of this molecule in patients as it becomes available on the market in different countries.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
