Doglas Parise, Mariana T D Parise, Marcus V C Viana, Adrian V Muñoz-Bucio, Yazmin A Cortés-Pérez, Beatriz Arellano-Reynoso, Efrén Díaz-Aparicio, Fernanda A Dorella, Felipe L Pereira, Alex F Carvalho, Henrique C P Figueiredo, Preetam Ghosh, Debmalya Barh, Anne C P Gomide, Vasco A C Azevedo
{"title":"First genome sequencing and comparative analyses of <i>Corynebacterium pseudotuberculosis</i> strains from Mexico.","authors":"Doglas Parise, Mariana T D Parise, Marcus V C Viana, Adrian V Muñoz-Bucio, Yazmin A Cortés-Pérez, Beatriz Arellano-Reynoso, Efrén Díaz-Aparicio, Fernanda A Dorella, Felipe L Pereira, Alex F Carvalho, Henrique C P Figueiredo, Preetam Ghosh, Debmalya Barh, Anne C P Gomide, Vasco A C Azevedo","doi":"10.1186/s40793-018-0325-z","DOIUrl":null,"url":null,"abstract":"<p><p><i>Corynebacterium pseudotuberculosis</i> is a pathogenic bacterium which has been rapidly spreading all over the world, causing economic losses in the agricultural sector and sporadically infecting humans. Six <i>C. pseudotuberculosis</i> strains were isolated from goats, sheep, and horses with distinct abscess locations. For the first time, Mexican genomes of this bacterium were sequenced and studied in silico. All strains were sequenced using Ion Personal Genome Machine sequencer, assembled using Newbler and SPAdes software. The automatic genome annotation was done using the software RAST and in-house scripts for transference, followed by manual curation using Artemis software and BLAST against NCBI and UniProt databases. The six genomes are publicly available in NCBI database. The analysis of nucleotide sequence similarity and the generated phylogenetic tree led to the observation that the Mexican strains are more similar between strains from the same host, but the genetic structure is probably more influenced by transportation of animals between farms than host preference. Also, a putative drug target was predicted and in silico analysis of 46 strains showed two gene clusters capable of differentiating the biovars <i>equi</i> and <i>ovis</i>: Restriction Modification system and CRISPR-Cas cluster.</p>","PeriodicalId":21965,"journal":{"name":"Standards in Genomic Sciences","volume":"13 ","pages":"21"},"PeriodicalIF":0.0000,"publicationDate":"2018-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6180578/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Standards in Genomic Sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40793-018-0325-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
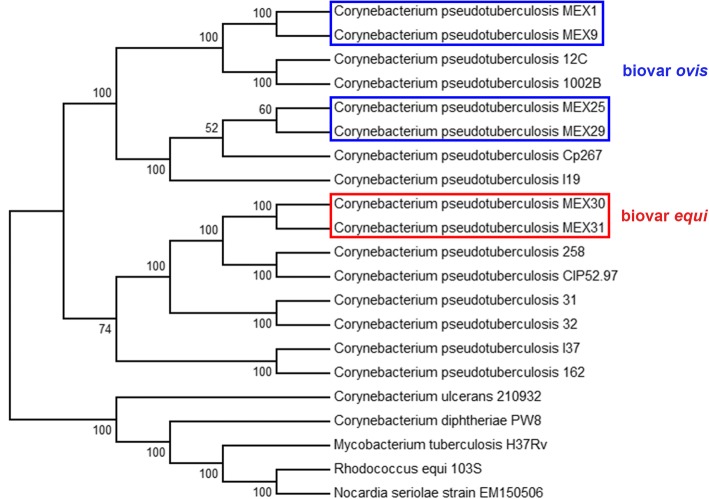
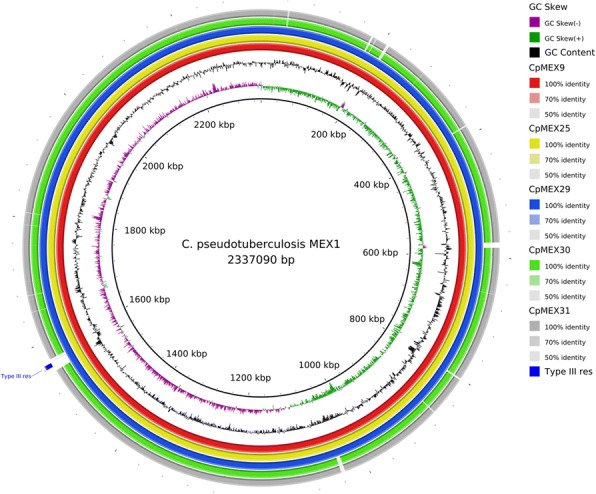
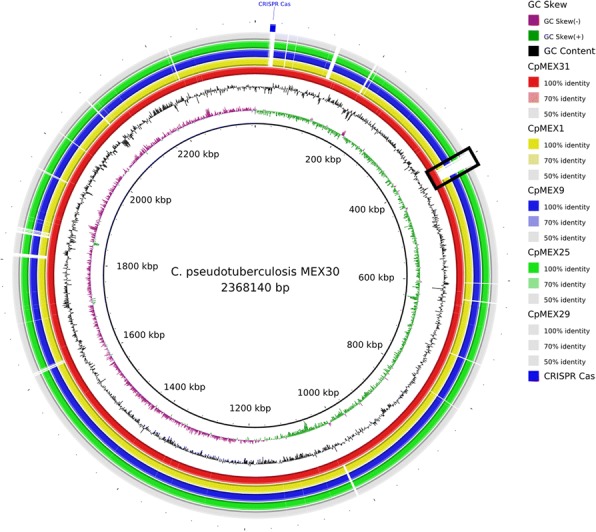
Corynebacterium pseudotuberculosis is a pathogenic bacterium which has been rapidly spreading all over the world, causing economic losses in the agricultural sector and sporadically infecting humans. Six C. pseudotuberculosis strains were isolated from goats, sheep, and horses with distinct abscess locations. For the first time, Mexican genomes of this bacterium were sequenced and studied in silico. All strains were sequenced using Ion Personal Genome Machine sequencer, assembled using Newbler and SPAdes software. The automatic genome annotation was done using the software RAST and in-house scripts for transference, followed by manual curation using Artemis software and BLAST against NCBI and UniProt databases. The six genomes are publicly available in NCBI database. The analysis of nucleotide sequence similarity and the generated phylogenetic tree led to the observation that the Mexican strains are more similar between strains from the same host, but the genetic structure is probably more influenced by transportation of animals between farms than host preference. Also, a putative drug target was predicted and in silico analysis of 46 strains showed two gene clusters capable of differentiating the biovars equi and ovis: Restriction Modification system and CRISPR-Cas cluster.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
