{"title":"Mitochondrial neurogastrointestinal encephalomyopathy: approaches to diagnosis and treatment.","authors":"Bridget E Bax","doi":"10.20517/jtgg.2020.08","DOIUrl":null,"url":null,"abstract":"<p><p>Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an ultra-rare disease caused by mutations in <i>TYMP,</i> the gene encoding for the enzyme thymidine phosphorylase. The resulting enzyme deficiency leads to a systemic accumulation of thymidine and 2'-deoxyuridine and ultimately mitochondrial failure due to a progressive acquisition of secondary mitochondrial DNA (mtDNA) mutations and mtDNA depletion. MNGIE is characterised by gastrointestinal dysmotility, cachexia, peripheral neuropathy, ophthalmoplegia, ptosis and leukoencephalopathy. The disease is progressively degenerative and leads to death at an average age of 37.6 years. Patients invariably encounter misdiagnoses, diagnostic delays, and non-specific clinical management. Despite its rarity, MNGIE has invoked much interest in the development of therapeutic strategies, mainly because it is one of the few mitochondrial disorders where the molecular abnormality is metabolically and physically accessible to manipulation. This review provides a resume of the current diagnosis and treatment approaches and aims to increase the clinical awareness of MNGIE and thereby facilitate early diagnosis and timely access to treatments, before the development of untreatable and irreversible organ damage.</p>","PeriodicalId":73999,"journal":{"name":"Journal of translational genetics and genomics","volume":"4 ","pages":"1-16"},"PeriodicalIF":1.0000,"publicationDate":"2020-03-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7116056/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of translational genetics and genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.20517/jtgg.2020.08","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
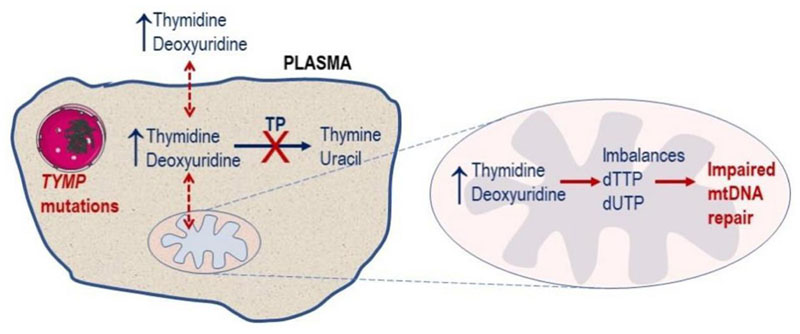
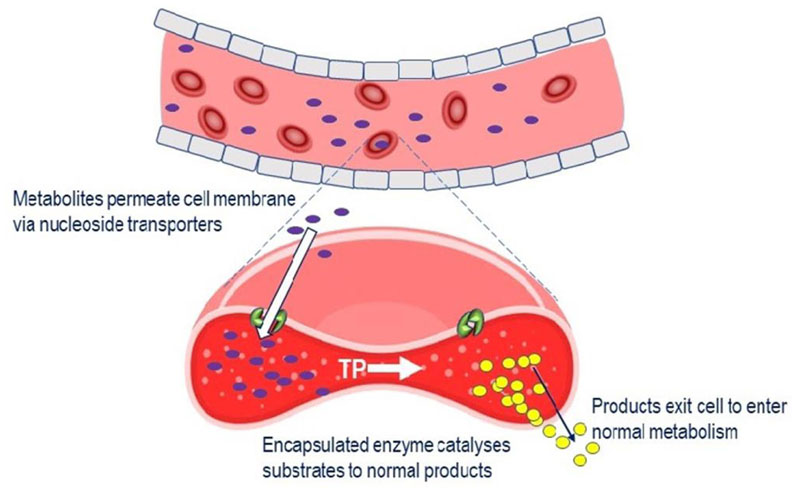
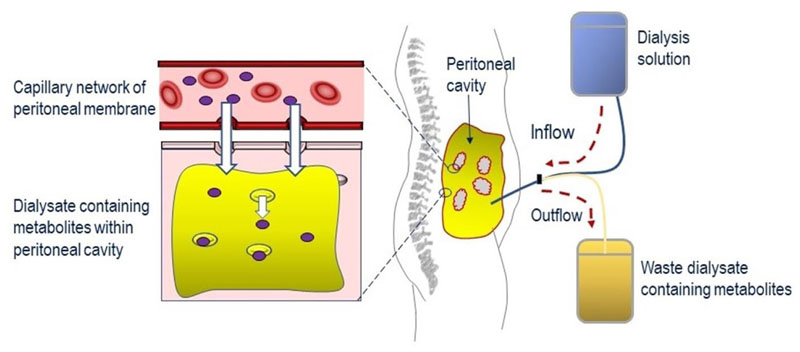
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an ultra-rare disease caused by mutations in TYMP, the gene encoding for the enzyme thymidine phosphorylase. The resulting enzyme deficiency leads to a systemic accumulation of thymidine and 2'-deoxyuridine and ultimately mitochondrial failure due to a progressive acquisition of secondary mitochondrial DNA (mtDNA) mutations and mtDNA depletion. MNGIE is characterised by gastrointestinal dysmotility, cachexia, peripheral neuropathy, ophthalmoplegia, ptosis and leukoencephalopathy. The disease is progressively degenerative and leads to death at an average age of 37.6 years. Patients invariably encounter misdiagnoses, diagnostic delays, and non-specific clinical management. Despite its rarity, MNGIE has invoked much interest in the development of therapeutic strategies, mainly because it is one of the few mitochondrial disorders where the molecular abnormality is metabolically and physically accessible to manipulation. This review provides a resume of the current diagnosis and treatment approaches and aims to increase the clinical awareness of MNGIE and thereby facilitate early diagnosis and timely access to treatments, before the development of untreatable and irreversible organ damage.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
