S T Nevin, N Lawrence, A Nicke, R J Lewis, D J Adams
{"title":"Functional modulation of the human voltage-gated sodium channel Na<sub>V</sub>1.8 by auxiliary β subunits.","authors":"S T Nevin, N Lawrence, A Nicke, R J Lewis, D J Adams","doi":"10.1080/19336950.2020.1860399","DOIUrl":null,"url":null,"abstract":"<p><p>The voltage-gated sodium channel Na<sub>v</sub>1.8 mediates the tetrodotoxin-resistant (TTX-R) Na<sup>+</sup> current in nociceptive primary sensory neurons, which has an important role in the transmission of painful stimuli. Here, we describe the functional modulation of the human Na<sub>v</sub>1.8 α-subunit in <i>Xenopus</i> oocytes by auxiliary β subunits. We found that the β3 subunit down-regulated the maximal Na<sup>+</sup> current amplitude and decelerated recovery from inactivation of hNa<sub>v</sub>1.8, whereas the β1 and β2 subunits had no such effects. The specific regulation of Na<sub>v</sub>1.8 by the β3 subunit constitutes a potential novel regulatory mechanism of the TTX-R Na<sup>+</sup> current in primary sensory neurons with potential implications in chronic pain states. In particular, neuropathic pain states are characterized by a down-regulation of Na<sub>v</sub>1.8 accompanied by increased expression of the β3 subunit. Our results suggest that these two phenomena may be correlated, and that increased levels of the β3 subunit may directly contribute to the down-regulation of Na<sub>v</sub>1.8. To determine which domain of the β3 subunit is responsible for the specific regulation of hNa<sub>v</sub>1.8, we created chimeras of the β1 and β3 subunits and co-expressed them with the hNa<sub>v</sub>1.8 α-subunit in <i>Xenopus</i> oocytes. The intracellular domain of the β3 subunit was shown to be responsible for the down-regulation of maximal Na<sub>v</sub>1.8 current amplitudes. In contrast, the extracellular domain mediated the effect of the β3 subunit on hNa<sub>v</sub>1.8 recovery kinetics.</p>","PeriodicalId":72555,"journal":{"name":"Channels (Austin, Tex.)","volume":" ","pages":"79-93"},"PeriodicalIF":3.2000,"publicationDate":"2021-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7781643/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Channels (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/19336950.2020.1860399","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
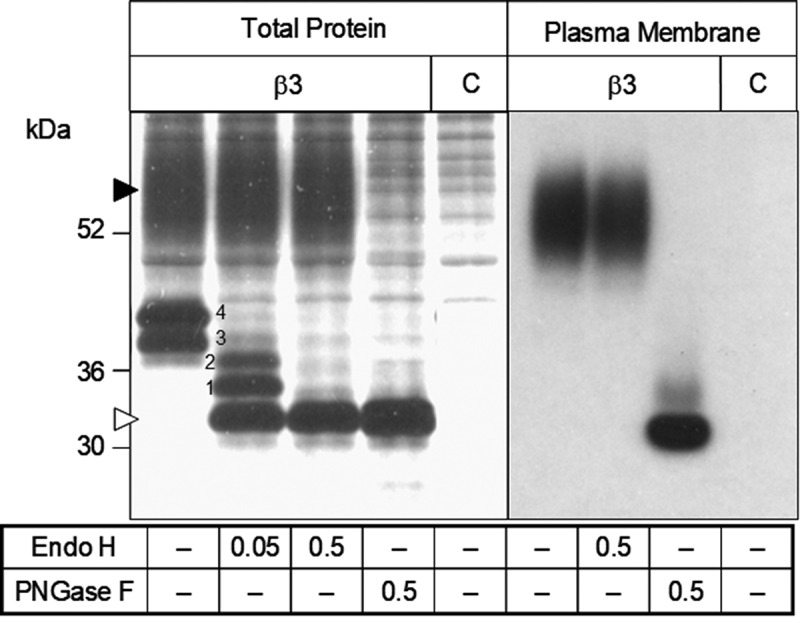
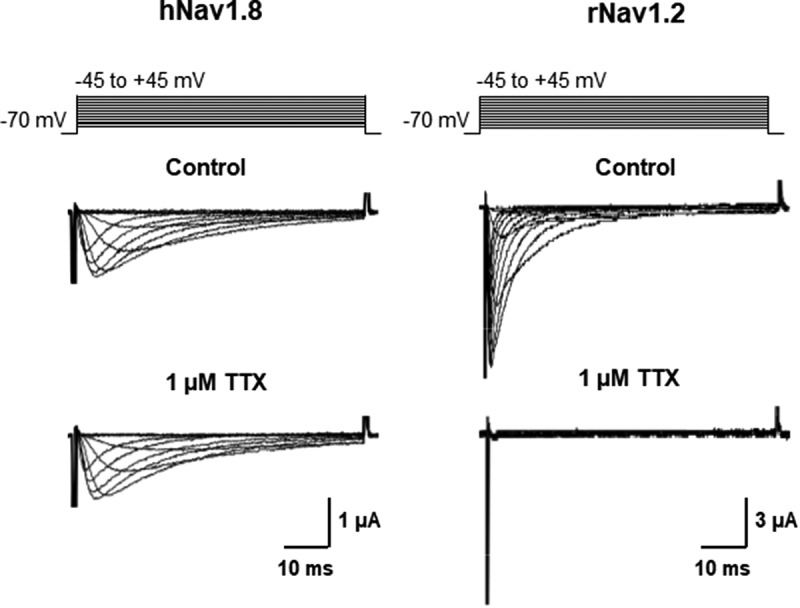
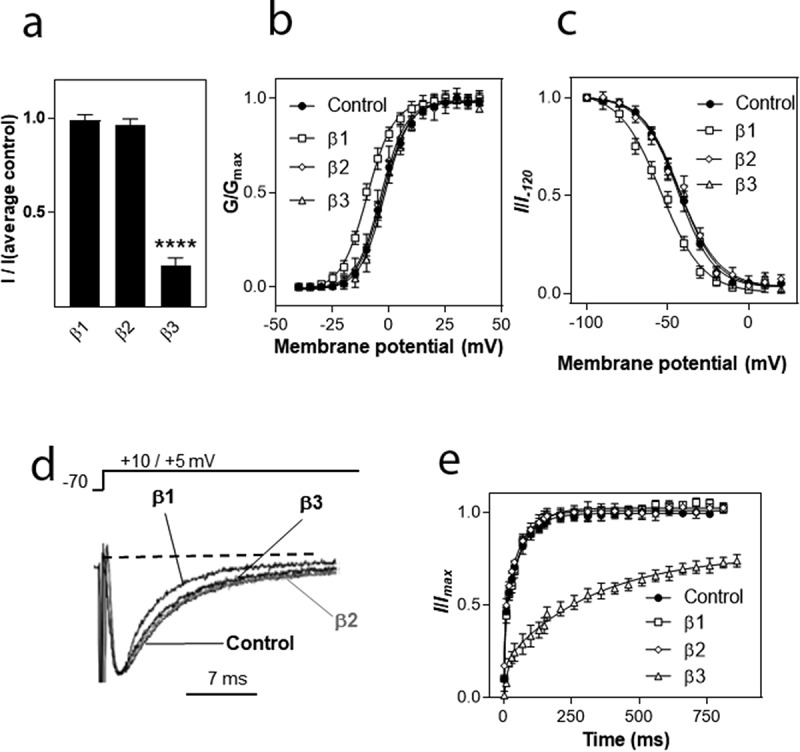
The voltage-gated sodium channel Nav1.8 mediates the tetrodotoxin-resistant (TTX-R) Na+ current in nociceptive primary sensory neurons, which has an important role in the transmission of painful stimuli. Here, we describe the functional modulation of the human Nav1.8 α-subunit in Xenopus oocytes by auxiliary β subunits. We found that the β3 subunit down-regulated the maximal Na+ current amplitude and decelerated recovery from inactivation of hNav1.8, whereas the β1 and β2 subunits had no such effects. The specific regulation of Nav1.8 by the β3 subunit constitutes a potential novel regulatory mechanism of the TTX-R Na+ current in primary sensory neurons with potential implications in chronic pain states. In particular, neuropathic pain states are characterized by a down-regulation of Nav1.8 accompanied by increased expression of the β3 subunit. Our results suggest that these two phenomena may be correlated, and that increased levels of the β3 subunit may directly contribute to the down-regulation of Nav1.8. To determine which domain of the β3 subunit is responsible for the specific regulation of hNav1.8, we created chimeras of the β1 and β3 subunits and co-expressed them with the hNav1.8 α-subunit in Xenopus oocytes. The intracellular domain of the β3 subunit was shown to be responsible for the down-regulation of maximal Nav1.8 current amplitudes. In contrast, the extracellular domain mediated the effect of the β3 subunit on hNav1.8 recovery kinetics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
