Vitaly A. Gorbunov, Anastasiia I. Uliankina and Alexander V. Myshlyavtsev
{"title":"Simple lattice model of surface-confined metal–organic networks consisting of linear nitrogen-bearing molecules and transition metals†","authors":"Vitaly A. Gorbunov, Anastasiia I. Uliankina and Alexander V. Myshlyavtsev","doi":"10.1039/D2ME00199C","DOIUrl":null,"url":null,"abstract":"<p >We propose a generalized lattice model that enables prediction of the phase behavior and thermal stability of surface-confined metal–organic layers consisting of molecules with nitrogen-bearing functional groups (–CN, –Py, (<img>NH)<small><sub>2</sub></small>) of various sizes and transition metal atoms (copper and iron). The coordination energy per molecule is revealed to be a nearly linear function of the coordination number. In the case of three-fold coordination and higher, steric repulsions between the coordinated functional groups play an important role. The lattice model has been parametrized using DFT methods. The ground state phase diagrams have been calculated and verified by GCMC simulation at non-zero temperatures. An increase in the size of the functional group and/or decrease of the coordination capacity of the metal center leads to a greater phase diversity. There are linear metal–organic structures and metal–organic networks consisting of different coordination motifs. Otherwise, close-packed structures with high coordination motifs predominate. The relative thermal stability of the linear and 2D porous metal–organic structures is proportional to the average coordination number of the structure. Thermal destruction of the porous metal–organic structures occurs through breaking the coordination bonds and compacting the layer at the first stage forming the motifs with higher coordination numbers.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 3","pages":" 349-357"},"PeriodicalIF":3.2000,"publicationDate":"2022-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/me/d2me00199c","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
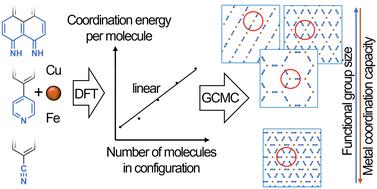
We propose a generalized lattice model that enables prediction of the phase behavior and thermal stability of surface-confined metal–organic layers consisting of molecules with nitrogen-bearing functional groups (–CN, –Py, (NH)2) of various sizes and transition metal atoms (copper and iron). The coordination energy per molecule is revealed to be a nearly linear function of the coordination number. In the case of three-fold coordination and higher, steric repulsions between the coordinated functional groups play an important role. The lattice model has been parametrized using DFT methods. The ground state phase diagrams have been calculated and verified by GCMC simulation at non-zero temperatures. An increase in the size of the functional group and/or decrease of the coordination capacity of the metal center leads to a greater phase diversity. There are linear metal–organic structures and metal–organic networks consisting of different coordination motifs. Otherwise, close-packed structures with high coordination motifs predominate. The relative thermal stability of the linear and 2D porous metal–organic structures is proportional to the average coordination number of the structure. Thermal destruction of the porous metal–organic structures occurs through breaking the coordination bonds and compacting the layer at the first stage forming the motifs with higher coordination numbers.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
