Vanessa E Jahnke, Jennifer M Peterson, Jack H Van Der Meulen, Jessica Boehler, Kitipong Uaesoontrachoon, Helen K Johnston, Aurelia Defour, Aditi Phadke, Qing Yu, Jyoti K Jaiswal, Kanneboyina Nagaraju
{"title":"Mitochondrial dysfunction and consequences in calpain-3-deficient muscle.","authors":"Vanessa E Jahnke, Jennifer M Peterson, Jack H Van Der Meulen, Jessica Boehler, Kitipong Uaesoontrachoon, Helen K Johnston, Aurelia Defour, Aditi Phadke, Qing Yu, Jyoti K Jaiswal, Kanneboyina Nagaraju","doi":"10.1186/s13395-020-00254-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Nonsense or loss-of-function mutations in the non-lysosomal cysteine protease calpain-3 result in limb-girdle muscular dystrophy type 2A (LGMD2A). While calpain-3 is implicated in muscle cell differentiation, sarcomere formation, and muscle cytoskeletal remodeling, the physiological basis for LGMD2A has remained elusive.</p><p><strong>Methods: </strong>Cell growth, gene expression profiling, and mitochondrial content and function were analyzed using muscle and muscle cell cultures established from healthy and calpain-3-deficient mice. Calpain-3-deficient mice were also treated with PPAR-delta agonist (GW501516) to assess mitochondrial function and membrane repair. The unpaired t test was used to assess the significance of the differences observed between the two groups or treatments. ANOVAs were used to assess significance over time.</p><p><strong>Results: </strong>We find that calpain-3 deficiency causes mitochondrial dysfunction in the muscles and myoblasts. Calpain-3-deficient myoblasts showed increased proliferation, and their gene expression profile showed aberrant mitochondrial biogenesis. Myotube gene expression analysis further revealed altered lipid metabolism in calpain-3-deficient muscle. Mitochondrial defects were validated in vitro and in vivo. We used GW501516 to improve mitochondrial biogenesis in vivo in 7-month-old calpain-3-deficient mice. This treatment improved satellite cell activity as indicated by increased MyoD and Pax7 mRNA expression. It also decreased muscle fatigability and reduced serum creatine kinase levels. The decreased mitochondrial function also impaired sarcolemmal repair in the calpain-3-deficient skeletal muscle. Improving mitochondrial activity by acute pyruvate treatment improved sarcolemmal repair.</p><p><strong>Conclusion: </strong>Our results provide evidence that calpain-3 deficiency in the skeletal muscle is associated with poor mitochondrial biogenesis and function resulting in poor sarcolemmal repair. Addressing this deficit by drugs that improve mitochondrial activity offers new therapeutic avenues for LGMD2A.</p>","PeriodicalId":5,"journal":{"name":"ACS Applied Materials & Interfaces","volume":" ","pages":"37"},"PeriodicalIF":8.2000,"publicationDate":"2020-12-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7730798/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Materials & Interfaces","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-020-00254-1","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Nonsense or loss-of-function mutations in the non-lysosomal cysteine protease calpain-3 result in limb-girdle muscular dystrophy type 2A (LGMD2A). While calpain-3 is implicated in muscle cell differentiation, sarcomere formation, and muscle cytoskeletal remodeling, the physiological basis for LGMD2A has remained elusive.
Methods: Cell growth, gene expression profiling, and mitochondrial content and function were analyzed using muscle and muscle cell cultures established from healthy and calpain-3-deficient mice. Calpain-3-deficient mice were also treated with PPAR-delta agonist (GW501516) to assess mitochondrial function and membrane repair. The unpaired t test was used to assess the significance of the differences observed between the two groups or treatments. ANOVAs were used to assess significance over time.
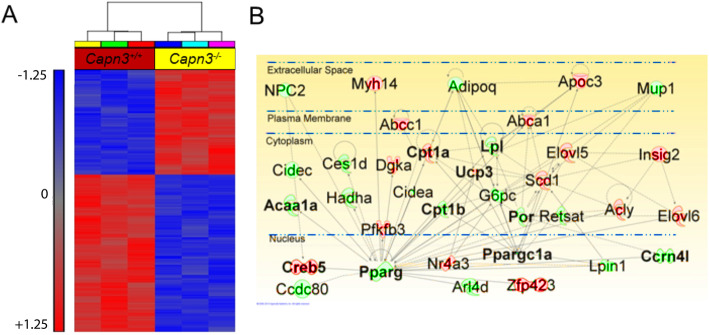
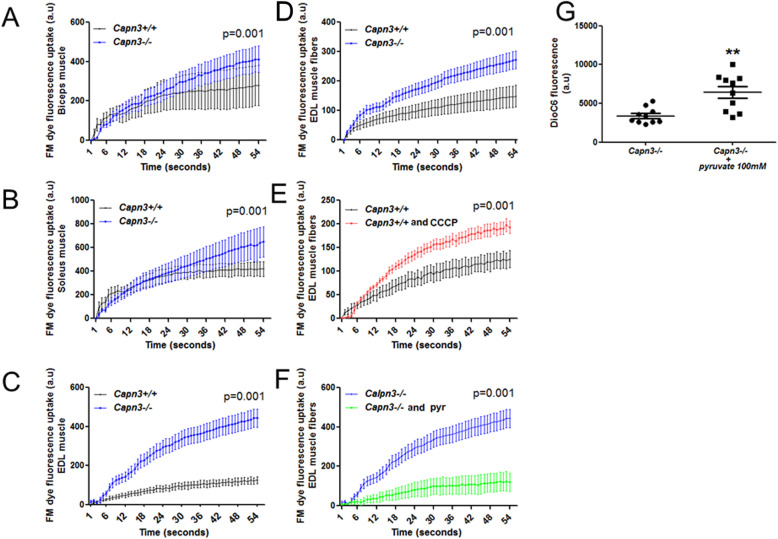
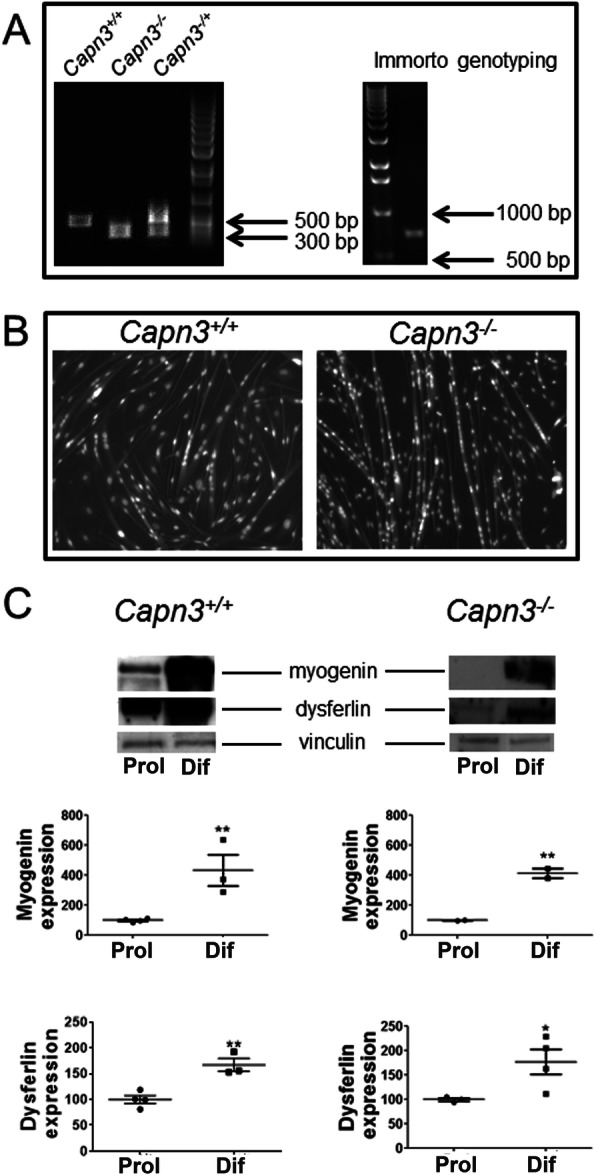
Results: We find that calpain-3 deficiency causes mitochondrial dysfunction in the muscles and myoblasts. Calpain-3-deficient myoblasts showed increased proliferation, and their gene expression profile showed aberrant mitochondrial biogenesis. Myotube gene expression analysis further revealed altered lipid metabolism in calpain-3-deficient muscle. Mitochondrial defects were validated in vitro and in vivo. We used GW501516 to improve mitochondrial biogenesis in vivo in 7-month-old calpain-3-deficient mice. This treatment improved satellite cell activity as indicated by increased MyoD and Pax7 mRNA expression. It also decreased muscle fatigability and reduced serum creatine kinase levels. The decreased mitochondrial function also impaired sarcolemmal repair in the calpain-3-deficient skeletal muscle. Improving mitochondrial activity by acute pyruvate treatment improved sarcolemmal repair.
Conclusion: Our results provide evidence that calpain-3 deficiency in the skeletal muscle is associated with poor mitochondrial biogenesis and function resulting in poor sarcolemmal repair. Addressing this deficit by drugs that improve mitochondrial activity offers new therapeutic avenues for LGMD2A.
期刊介绍:
ACS Applied Materials & Interfaces is a leading interdisciplinary journal that brings together chemists, engineers, physicists, and biologists to explore the development and utilization of newly-discovered materials and interfacial processes for specific applications. Our journal has experienced remarkable growth since its establishment in 2009, both in terms of the number of articles published and the impact of the research showcased. We are proud to foster a truly global community, with the majority of published articles originating from outside the United States, reflecting the rapid growth of applied research worldwide.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
