{"title":"Redefining infantile-onset multisystem phenotypes of coenzyme Q<sub>10</sub>-deficiency in the next-generation sequencing era.","authors":"Andres Berardo, Catarina M Quinzii","doi":"10.20517/jtgg.2020.02","DOIUrl":null,"url":null,"abstract":"<p><p>Primary coenzyme Q<sub>10</sub> (CoQ<sub>10</sub>) deficiency encompasses a subset of mitochondrial diseases caused by mutations affecting proteins involved in the CoQ<sub>10</sub> biosynthetic pathway. One of the most frequent clinical syndromes associated with primary CoQ<sub>10</sub> deficiency is the severe infantile multisystemic form, which, until recently, was underdiagnosed. In the last few years, the availability of genetic screening through whole exome sequencing and whole genome sequencing has enabled molecular diagnosis in a growing number of patients with this syndrome and has revealed new disease phenotypes and molecular defects in CoQ<sub>10</sub> biosynthetic pathway genes. Early genetic screening can rapidly and non-invasively diagnose primary CoQ<sub>10</sub> deficiencies. Early diagnosis is particularly important in cases of CoQ<sub>10</sub> deficient steroid-resistant nephrotic syndrome, which frequently improves with treatment. In contrast, the infantile multisystemic forms of CoQ<sub>10</sub> deficiency, particularly when manifesting with encephalopathy, present therapeutic challenges, due to poor responses to CoQ<sub>10</sub> supplementation. Administration of CoQ<sub>10</sub> biosynthetic intermediate compounds is a promising alternative to CoQ<sub>10</sub>; however, further pre-clinical studies are needed to establish their safety and efficacy, as well as to elucidate the mechanism of actions of the intermediates. Here, we review the molecular defects causes of the multisystemic infantile phenotype of primary CoQ<sub>10</sub> deficiency, genotype-phenotype correlations, and recent therapeutic advances.</p>","PeriodicalId":73999,"journal":{"name":"Journal of translational genetics and genomics","volume":"4 ","pages":"22-35"},"PeriodicalIF":1.0000,"publicationDate":"2020-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7791541/pdf/","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of translational genetics and genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.20517/jtgg.2020.02","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/4/23 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 7
Abstract
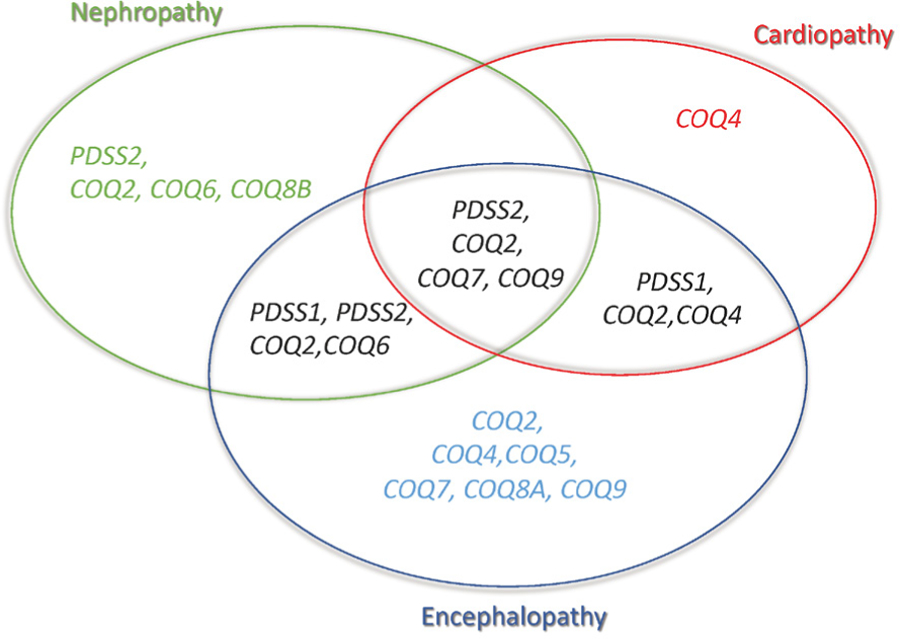
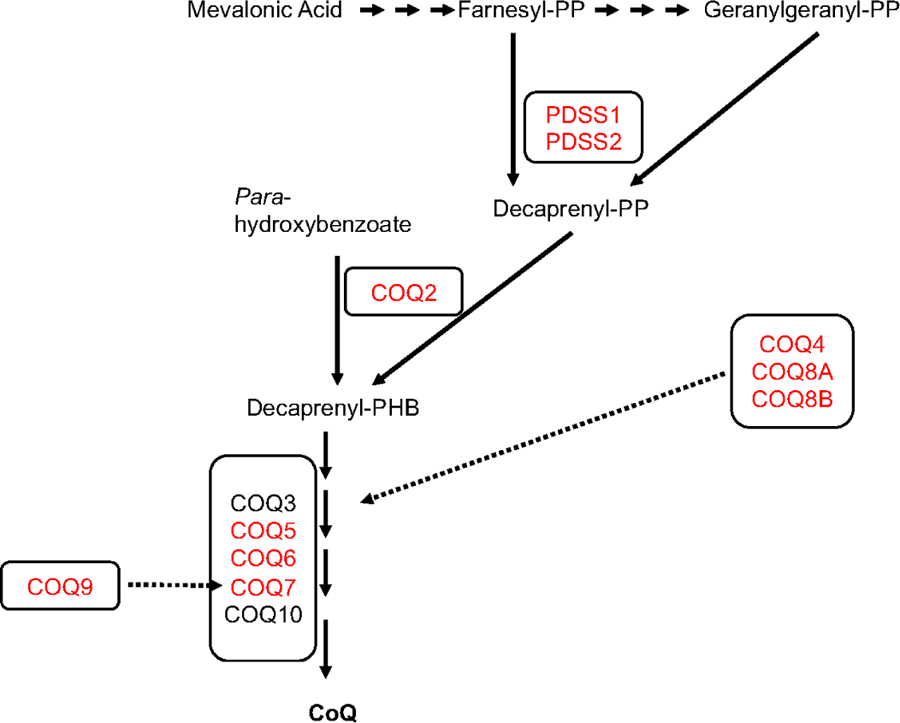
Primary coenzyme Q10 (CoQ10) deficiency encompasses a subset of mitochondrial diseases caused by mutations affecting proteins involved in the CoQ10 biosynthetic pathway. One of the most frequent clinical syndromes associated with primary CoQ10 deficiency is the severe infantile multisystemic form, which, until recently, was underdiagnosed. In the last few years, the availability of genetic screening through whole exome sequencing and whole genome sequencing has enabled molecular diagnosis in a growing number of patients with this syndrome and has revealed new disease phenotypes and molecular defects in CoQ10 biosynthetic pathway genes. Early genetic screening can rapidly and non-invasively diagnose primary CoQ10 deficiencies. Early diagnosis is particularly important in cases of CoQ10 deficient steroid-resistant nephrotic syndrome, which frequently improves with treatment. In contrast, the infantile multisystemic forms of CoQ10 deficiency, particularly when manifesting with encephalopathy, present therapeutic challenges, due to poor responses to CoQ10 supplementation. Administration of CoQ10 biosynthetic intermediate compounds is a promising alternative to CoQ10; however, further pre-clinical studies are needed to establish their safety and efficacy, as well as to elucidate the mechanism of actions of the intermediates. Here, we review the molecular defects causes of the multisystemic infantile phenotype of primary CoQ10 deficiency, genotype-phenotype correlations, and recent therapeutic advances.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
