Rebeca García-Fandiño, Moisés Gulías, Jose L. Mascareñas and Diego J. Cárdenas
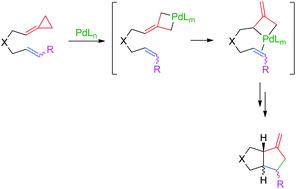
{"title":"Mechanistic study on the palladium-catalyzed (3 + 2) intramolecular cycloaddition of alk-5-enylidenecyclopropanes†","authors":"Rebeca García-Fandiño, Moisés Gulías, Jose L. Mascareñas and Diego J. Cárdenas","doi":"10.1039/C2DT11955B","DOIUrl":null,"url":null,"abstract":"<p >The intramolecular (3 + 2) <annref>cycloaddition</annref> of alkenylidenecyclopropanes to <annref>alkenes</annref> under palladium catalysis provides a practical and stereoselective entry into a variety of interesting bicycles. The reaction outcome and stereoselectivity of the process are somewhat dependent on the characteristics of the substrate and of the <annref>palladium</annref> <annref>ligand</annref>, which is not easy to justify on the basis of the current mechanistic understanding. We therefore decided to study the different mechanistic alternatives from a theoretical point of view. The energies of the <annref>reaction intermediates</annref> and transition states for different possible pathways have been explored at DFT level in a model system, and using PH<small><sub>3</sub></small> and P(OMe)<small><sub>3</sub></small> as <annref>ligands</annref>. The results obtained suggest that the most favourable reaction pathway involves an initial oxidative addition of Pd(0) at the distal position of the <annref>cyclopropane</annref> to afford a palladacyclobutane intermediate. The evolution of this intermediate into the final cycloadduct can occur following different paths, the most favorable depending on the configuration and substitution of the <annref>alkene</annref> cycloaddition partner, and the number of ancillary <annref>ligands</annref> coordinated to Pd. The computational results are consistent with the experimental observations and provide the basis for proposing which would be the operative mechanistic pathway in different cases. The results also allow us to explain the stereochemical divergences observed in some of the reactions.</p>","PeriodicalId":71,"journal":{"name":"Dalton Transactions","volume":" 31","pages":" 9468-9481"},"PeriodicalIF":3.3000,"publicationDate":"2012-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C2DT11955B","citationCount":"15","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Dalton Transactions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2012/dt/c2dt11955b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 15
Abstract
The intramolecular (3 + 2) cycloaddition of alkenylidenecyclopropanes to alkenes under palladium catalysis provides a practical and stereoselective entry into a variety of interesting bicycles. The reaction outcome and stereoselectivity of the process are somewhat dependent on the characteristics of the substrate and of the palladiumligand, which is not easy to justify on the basis of the current mechanistic understanding. We therefore decided to study the different mechanistic alternatives from a theoretical point of view. The energies of the reaction intermediates and transition states for different possible pathways have been explored at DFT level in a model system, and using PH3 and P(OMe)3 as ligands. The results obtained suggest that the most favourable reaction pathway involves an initial oxidative addition of Pd(0) at the distal position of the cyclopropane to afford a palladacyclobutane intermediate. The evolution of this intermediate into the final cycloadduct can occur following different paths, the most favorable depending on the configuration and substitution of the alkene cycloaddition partner, and the number of ancillary ligands coordinated to Pd. The computational results are consistent with the experimental observations and provide the basis for proposing which would be the operative mechanistic pathway in different cases. The results also allow us to explain the stereochemical divergences observed in some of the reactions.
期刊介绍:
Dalton Transactions is a journal for all areas of inorganic chemistry, which encompasses the organometallic, bioinorganic and materials chemistry of the elements, with applications including synthesis, catalysis, energy conversion/storage, electrical devices and medicine. Dalton Transactions welcomes high-quality, original submissions in all of these areas and more, where the advancement of knowledge in inorganic chemistry is significant.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
