Integrating single cell sequencing with a spatial quantitative systems pharmacology model spQSP for personalized prediction of triple-negative breast cancer immunotherapy response
Shuming Zhang , Chang Gong , Alvaro Ruiz-Martinez , Hanwen Wang , Emily Davis-Marcisak , Atul Deshpande , Aleksander S. Popel , Elana J. Fertig
{"title":"Integrating single cell sequencing with a spatial quantitative systems pharmacology model spQSP for personalized prediction of triple-negative breast cancer immunotherapy response","authors":"Shuming Zhang , Chang Gong , Alvaro Ruiz-Martinez , Hanwen Wang , Emily Davis-Marcisak , Atul Deshpande , Aleksander S. Popel , Elana J. Fertig","doi":"10.1016/j.immuno.2021.100002","DOIUrl":null,"url":null,"abstract":"<div><p>Response to cancer immunotherapies depends on the complex and dynamic interactions between T cell recognition and killing of cancer cells that are counteracted through immunosuppressive pathways in the tumor microenvironment. Therefore, while measurements such as tumor mutational burden provide biomarkers to select patients for immunotherapy, they neither universally predict patient response nor implicate the mechanisms that underlie immunotherapy resistance. Recent advances in single-cell RNA sequencing technology measure cellular heterogeneity within cells of an individual tumor but have yet to realize the promise of predictive oncology. In addition to data, mechanistic multiscale computational models are developed to predict treatment response. Incorporating single-cell data from tumors to parameterize these computational models provides deeper insights into prediction of clinical outcome in individual patients. Here, we integrate whole-exome sequencing and scRNA-seq data from Triple-Negative Breast Cancer patients to model neoantigen burden in tumor cells as input to a spatial Quantitative System Pharmacology model. The model comprises a four-compartmental Quantitative System Pharmacology sub-model to represent a whole patient and a spatial agent-based sub-model to represent tumor volumes at the cellular scale. We use the high-throughput single-cell data to model the role of antigen burden and heterogeneity relative to the tumor microenvironment composition on predicted immunotherapy response. We demonstrate how this integrated modeling and single-cell analysis framework can be used to relate neoantigen heterogeneity to immunotherapy treatment outcomes. Our results demonstrate feasibility of merging single-cell data to initialize cell states in multiscale computational models such as the spQSP for personalized prediction of clinical outcomes to immunotherapy.</p></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"1 ","pages":"Article 100002"},"PeriodicalIF":0.0000,"publicationDate":"2021-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.immuno.2021.100002","citationCount":"17","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119021000021","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 17
Abstract
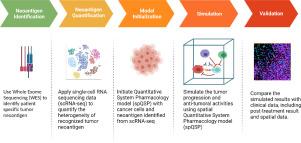
Response to cancer immunotherapies depends on the complex and dynamic interactions between T cell recognition and killing of cancer cells that are counteracted through immunosuppressive pathways in the tumor microenvironment. Therefore, while measurements such as tumor mutational burden provide biomarkers to select patients for immunotherapy, they neither universally predict patient response nor implicate the mechanisms that underlie immunotherapy resistance. Recent advances in single-cell RNA sequencing technology measure cellular heterogeneity within cells of an individual tumor but have yet to realize the promise of predictive oncology. In addition to data, mechanistic multiscale computational models are developed to predict treatment response. Incorporating single-cell data from tumors to parameterize these computational models provides deeper insights into prediction of clinical outcome in individual patients. Here, we integrate whole-exome sequencing and scRNA-seq data from Triple-Negative Breast Cancer patients to model neoantigen burden in tumor cells as input to a spatial Quantitative System Pharmacology model. The model comprises a four-compartmental Quantitative System Pharmacology sub-model to represent a whole patient and a spatial agent-based sub-model to represent tumor volumes at the cellular scale. We use the high-throughput single-cell data to model the role of antigen burden and heterogeneity relative to the tumor microenvironment composition on predicted immunotherapy response. We demonstrate how this integrated modeling and single-cell analysis framework can be used to relate neoantigen heterogeneity to immunotherapy treatment outcomes. Our results demonstrate feasibility of merging single-cell data to initialize cell states in multiscale computational models such as the spQSP for personalized prediction of clinical outcomes to immunotherapy.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
