{"title":"Synthaser: a CD-Search enabled Python toolkit for analysing domain architecture of fungal secondary metabolite megasynth(et)ases.","authors":"Cameron L M Gilchrist, Yit-Heng Chooi","doi":"10.1186/s40694-021-00120-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Fungi are prolific producers of secondary metabolites (SMs), which are bioactive small molecules with important applications in medicine, agriculture and other industries. The backbones of a large proportion of fungal SMs are generated through the action of large, multi-domain megasynth(et)ases such as polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs). The structure of these backbones is determined by the domain architecture of the corresponding megasynth(et)ase, and thus accurate annotation and classification of these architectures is an important step in linking SMs to their biosynthetic origins in the genome.</p><p><strong>Results: </strong>Here we report synthaser, a Python package leveraging the NCBI's conserved domain search tool for remote prediction and classification of fungal megasynth(et)ase domain architectures. Synthaser is capable of batch sequence analysis, and produces rich textual output and interactive visualisations which allow for quick assessment of the megasynth(et)ase diversity of a fungal genome. Synthaser uses a hierarchical rule-based classification system, which can be extensively customised by the user through a web application ( http://gamcil.github.io/synthaser ). We show that synthaser provides more accurate domain architecture predictions than comparable tools which rely on curated profile hidden Markov model (pHMM)-based approaches; the utilisation of the NCBI conserved domain database also allows for significantly greater flexibility compared to pHMM approaches. In addition, we demonstrate how synthaser can be applied to large scale genome mining pipelines through the construction of an Aspergillus PKS similarity network.</p><p><strong>Conclusions: </strong>Synthaser is an easy to use tool that represents a significant upgrade to previous domain architecture analysis tools. It is freely available under a MIT license from PyPI ( https://pypi.org/project/synthaser ) and GitHub ( https://github.com/gamcil/synthaser ).</p>","PeriodicalId":52292,"journal":{"name":"Fungal Biology and Biotechnology","volume":"8 1","pages":"13"},"PeriodicalIF":0.0000,"publicationDate":"2021-11-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8582187/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Fungal Biology and Biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40694-021-00120-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Fungi are prolific producers of secondary metabolites (SMs), which are bioactive small molecules with important applications in medicine, agriculture and other industries. The backbones of a large proportion of fungal SMs are generated through the action of large, multi-domain megasynth(et)ases such as polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs). The structure of these backbones is determined by the domain architecture of the corresponding megasynth(et)ase, and thus accurate annotation and classification of these architectures is an important step in linking SMs to their biosynthetic origins in the genome.
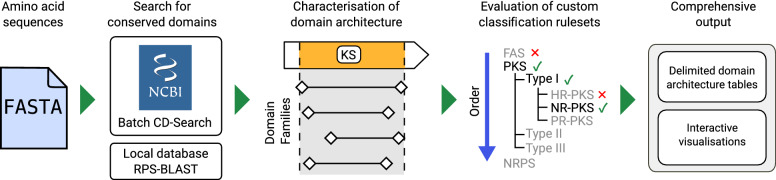
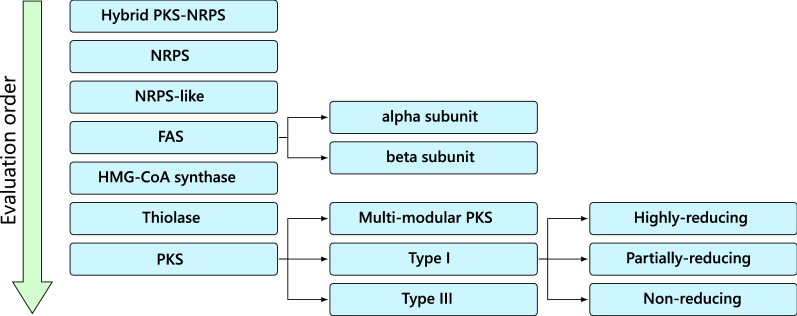
Results: Here we report synthaser, a Python package leveraging the NCBI's conserved domain search tool for remote prediction and classification of fungal megasynth(et)ase domain architectures. Synthaser is capable of batch sequence analysis, and produces rich textual output and interactive visualisations which allow for quick assessment of the megasynth(et)ase diversity of a fungal genome. Synthaser uses a hierarchical rule-based classification system, which can be extensively customised by the user through a web application ( http://gamcil.github.io/synthaser ). We show that synthaser provides more accurate domain architecture predictions than comparable tools which rely on curated profile hidden Markov model (pHMM)-based approaches; the utilisation of the NCBI conserved domain database also allows for significantly greater flexibility compared to pHMM approaches. In addition, we demonstrate how synthaser can be applied to large scale genome mining pipelines through the construction of an Aspergillus PKS similarity network.
Conclusions: Synthaser is an easy to use tool that represents a significant upgrade to previous domain architecture analysis tools. It is freely available under a MIT license from PyPI ( https://pypi.org/project/synthaser ) and GitHub ( https://github.com/gamcil/synthaser ).




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
