Linfang Wang, Honglei Wang, Shuanglong Yi, Shiping Zhang, Margaret S Ho
{"title":"A LRRK2/dLRRK-mediated lysosomal pathway that contributes to glial cell death and DA neuron survival.","authors":"Linfang Wang, Honglei Wang, Shuanglong Yi, Shiping Zhang, Margaret S Ho","doi":"10.1111/tra.12866","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of familial and sporadic Parkinson's disease. A plethora of evidence has indicated a role for LRRK2 in endolysosomal trafficking in neurons, while LRRK2 function in glia, although highly expressed, remains largely unknown. Here, we present evidence that LRRK2/dLRRK mediates a lysosomal pathway that contributes to glial cell death and the survival of dopaminergic (DA) neurons. LRRK2/dLRRK knockdown in the immortalized microglia or flies results in enlarged and swelling lysosomes fewer in number. These lysosomes are less mobile, wrongly acidified, exhibit defective membrane permeability and reduced activity of the lysosome hydrolase cathepsin B. In addition, LRRK2/dLRRK depletion causes glial apoptosis, DA neurodegeneration, and locomotor deficits in an age-dependent manner. Taken together, these findings demonstrate a functional role of LRRK2/dLRRK in regulating the glial lysosomal pathway; deficits in lysosomal biogenesis and function linking to glial apoptosis potentially underlie the mechanism of DA neurodegeneration, providing insights on LRRK2/dLRRK function in normal and pathological brains.</p>","PeriodicalId":427963,"journal":{"name":"Traffic (Copenhagen, Denmark)","volume":" ","pages":"506-520"},"PeriodicalIF":0.0000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Traffic (Copenhagen, Denmark)","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1111/tra.12866","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/11 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
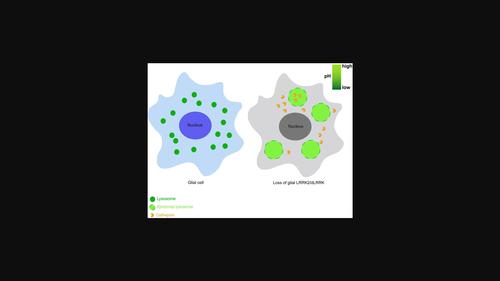
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of familial and sporadic Parkinson's disease. A plethora of evidence has indicated a role for LRRK2 in endolysosomal trafficking in neurons, while LRRK2 function in glia, although highly expressed, remains largely unknown. Here, we present evidence that LRRK2/dLRRK mediates a lysosomal pathway that contributes to glial cell death and the survival of dopaminergic (DA) neurons. LRRK2/dLRRK knockdown in the immortalized microglia or flies results in enlarged and swelling lysosomes fewer in number. These lysosomes are less mobile, wrongly acidified, exhibit defective membrane permeability and reduced activity of the lysosome hydrolase cathepsin B. In addition, LRRK2/dLRRK depletion causes glial apoptosis, DA neurodegeneration, and locomotor deficits in an age-dependent manner. Taken together, these findings demonstrate a functional role of LRRK2/dLRRK in regulating the glial lysosomal pathway; deficits in lysosomal biogenesis and function linking to glial apoptosis potentially underlie the mechanism of DA neurodegeneration, providing insights on LRRK2/dLRRK function in normal and pathological brains.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
