Francesca Magri, Sara Antognozzi, Michela Ripolone, Simona Zanotti, Laura Napoli, Patrizia Ciscato, Daniele Velardo, Giulietta Scuvera, Valeria Nicotra, Antonella Giacobbe, Donatella Milani, Francesco Fortunato, Manuela Garbellini, Monica Sciacco, Stefania Corti, Giacomo Pietro Comi, Dario Ronchi
{"title":"Megaconial congenital muscular dystrophy due to novel CHKB variants: a case report and literature review.","authors":"Francesca Magri, Sara Antognozzi, Michela Ripolone, Simona Zanotti, Laura Napoli, Patrizia Ciscato, Daniele Velardo, Giulietta Scuvera, Valeria Nicotra, Antonella Giacobbe, Donatella Milani, Francesco Fortunato, Manuela Garbellini, Monica Sciacco, Stefania Corti, Giacomo Pietro Comi, Dario Ronchi","doi":"10.1186/s13395-022-00306-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Choline kinase beta (CHKB) catalyzes the first step in the de novo biosynthesis of phosphatidyl choline and phosphatidylethanolamine via the Kennedy pathway. Derangement of this pathway might also influence the homeostasis of mitochondrial membranes. Autosomal recessive CHKB mutations cause a rare form of congenital muscular dystrophy known as megaconial congenital muscular dystrophy (MCMD).</p><p><strong>Case presentation: </strong>We describe a novel proband presenting MCMD due to unpublished CHKB mutations. The patient is a 6-year-old boy who came to our attention for cognitive impairment and slowly progressive muscular weakness. He was the first son of non-consanguineous healthy parents from Sri Lanka. Neurological examination showed proximal weakness at four limbs, weak osteotendinous reflexes, Gowers' maneuver, and waddling gate. Creatine kinase levels were mildly increased. EMG and brain MRI were normal. Left quadriceps skeletal muscle biopsy showed a myopathic pattern with nuclear centralizations and connective tissue increase. Histological and histochemical staining suggested subsarcolemmal localization and dimensional increase of mitochondria. Ultrastructural analysis confirmed the presence of enlarged (\"megaconial\") mitochondria. Direct sequencing of CHKB identified two novel defects: the c.1060G > C (p.Gly354Arg) substitution and the c.448-56_29del intronic deletion, segregating from father and mother, respectively. Subcloning of RT-PCR amplicons from patient's muscle RNA showed that c.448-56_29del results in the partial retention (14 nucleotides) of intron 3, altering physiological splicing and transcript stability. Biochemical studies showed reduced levels of the mitochondrial fission factor DRP1 and the severe impairment of mitochondrial respiratory chain activity in patient's muscle compared to controls.</p><p><strong>Conclusions: </strong>This report expands the molecular findings associated with MCMD and confirms the importance of considering CHKB variants in the differential diagnosis of patients presenting with muscular dystrophy and mental retardation. The clinical outcome of MCMD patients seems to be influenced by CHKB molecular defects. Histological and ultrastructural examination of muscle biopsy directed molecular studies and allowed the identification and characterization of an intronic mutation, usually escaping standard molecular testing.</p>","PeriodicalId":21747,"journal":{"name":"Skeletal Muscle","volume":" ","pages":"23"},"PeriodicalIF":4.4000,"publicationDate":"2022-09-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9524117/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Skeletal Muscle","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-022-00306-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
Background: Choline kinase beta (CHKB) catalyzes the first step in the de novo biosynthesis of phosphatidyl choline and phosphatidylethanolamine via the Kennedy pathway. Derangement of this pathway might also influence the homeostasis of mitochondrial membranes. Autosomal recessive CHKB mutations cause a rare form of congenital muscular dystrophy known as megaconial congenital muscular dystrophy (MCMD).
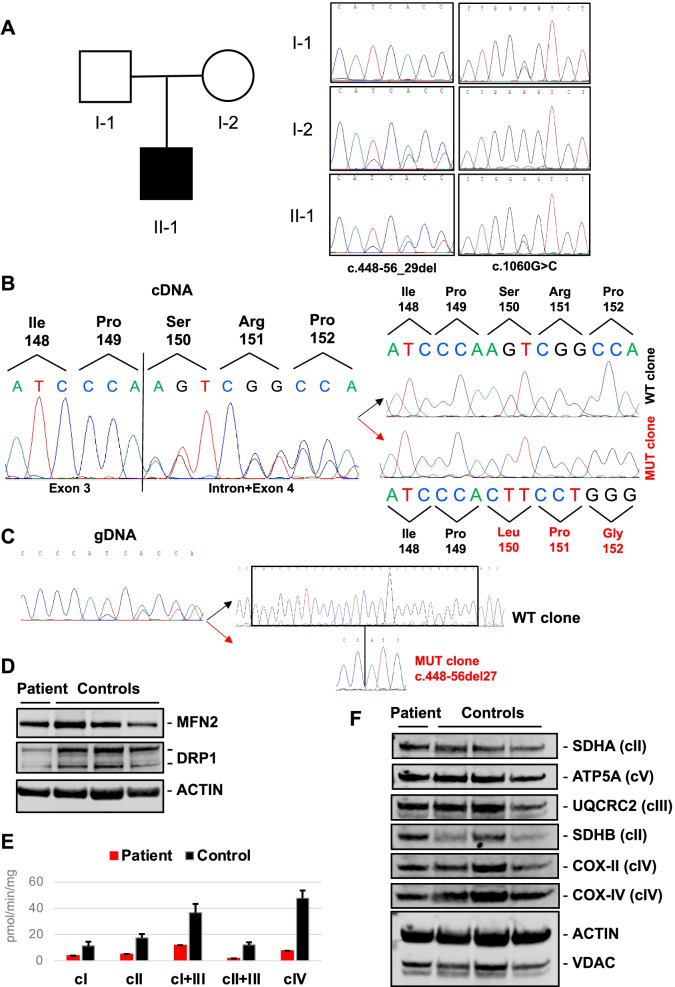
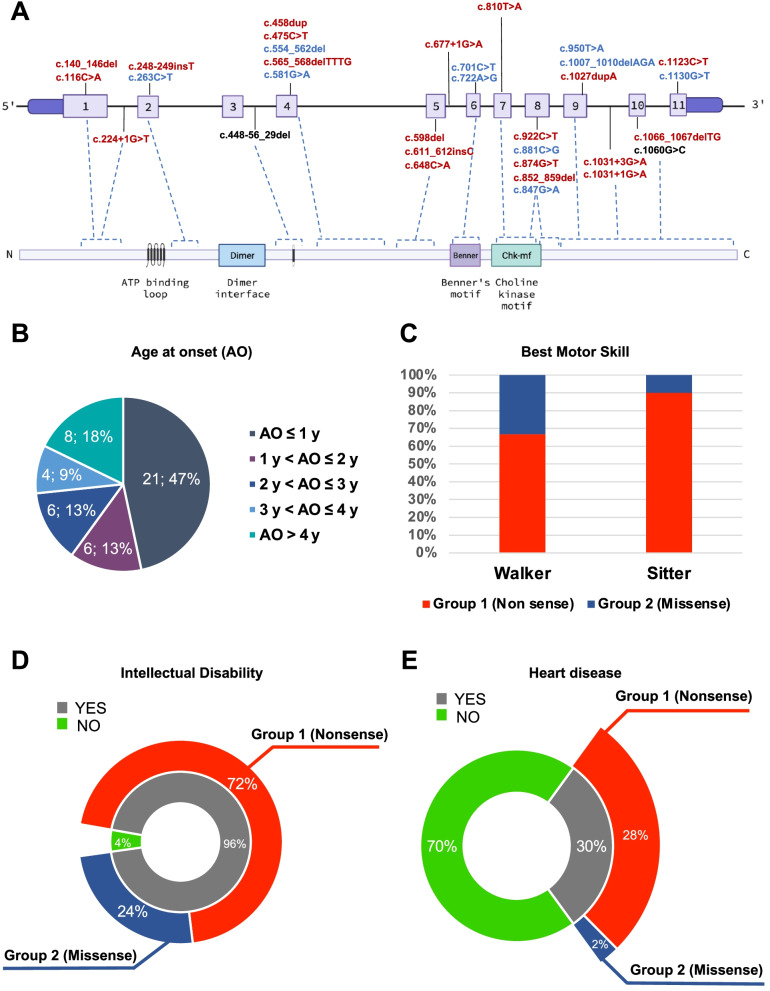
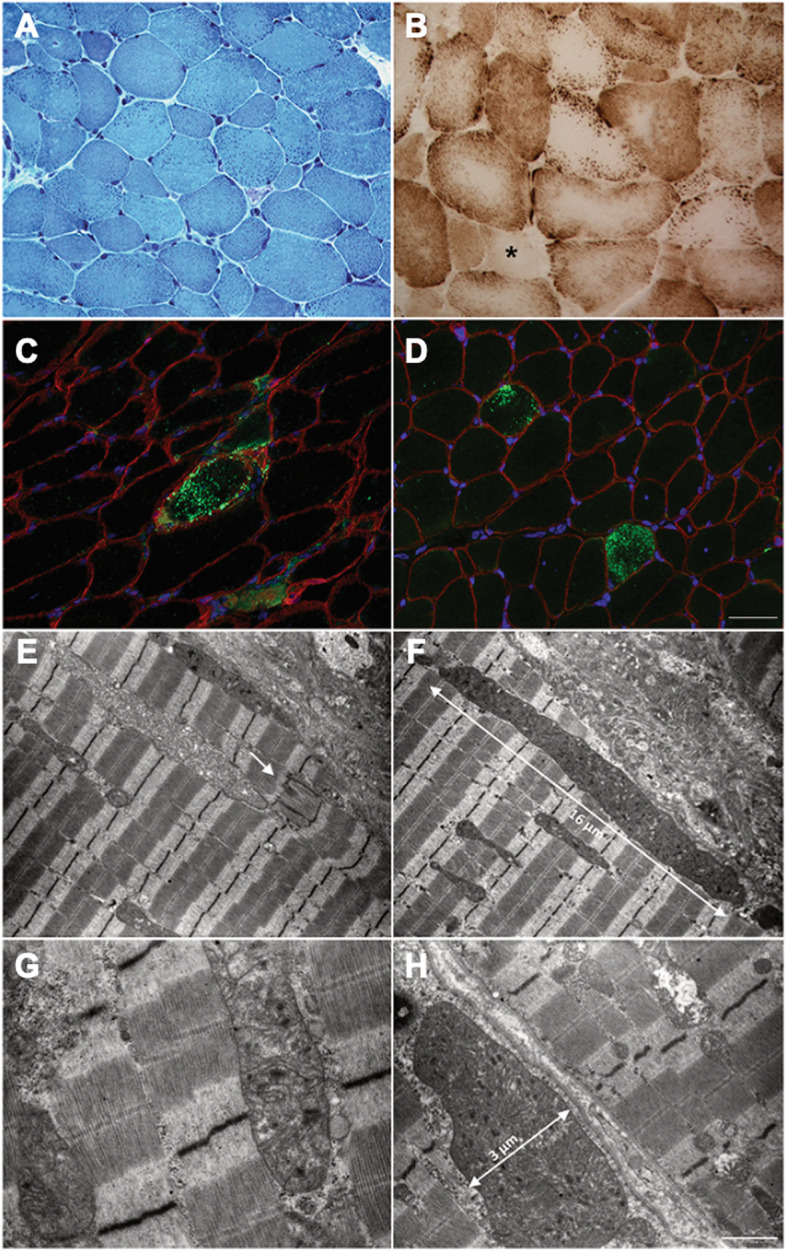
Case presentation: We describe a novel proband presenting MCMD due to unpublished CHKB mutations. The patient is a 6-year-old boy who came to our attention for cognitive impairment and slowly progressive muscular weakness. He was the first son of non-consanguineous healthy parents from Sri Lanka. Neurological examination showed proximal weakness at four limbs, weak osteotendinous reflexes, Gowers' maneuver, and waddling gate. Creatine kinase levels were mildly increased. EMG and brain MRI were normal. Left quadriceps skeletal muscle biopsy showed a myopathic pattern with nuclear centralizations and connective tissue increase. Histological and histochemical staining suggested subsarcolemmal localization and dimensional increase of mitochondria. Ultrastructural analysis confirmed the presence of enlarged ("megaconial") mitochondria. Direct sequencing of CHKB identified two novel defects: the c.1060G > C (p.Gly354Arg) substitution and the c.448-56_29del intronic deletion, segregating from father and mother, respectively. Subcloning of RT-PCR amplicons from patient's muscle RNA showed that c.448-56_29del results in the partial retention (14 nucleotides) of intron 3, altering physiological splicing and transcript stability. Biochemical studies showed reduced levels of the mitochondrial fission factor DRP1 and the severe impairment of mitochondrial respiratory chain activity in patient's muscle compared to controls.
Conclusions: This report expands the molecular findings associated with MCMD and confirms the importance of considering CHKB variants in the differential diagnosis of patients presenting with muscular dystrophy and mental retardation. The clinical outcome of MCMD patients seems to be influenced by CHKB molecular defects. Histological and ultrastructural examination of muscle biopsy directed molecular studies and allowed the identification and characterization of an intronic mutation, usually escaping standard molecular testing.
期刊介绍:
The only open access journal in its field, Skeletal Muscle publishes novel, cutting-edge research and technological advancements that investigate the molecular mechanisms underlying the biology of skeletal muscle. Reflecting the breadth of research in this area, the journal welcomes manuscripts about the development, metabolism, the regulation of mass and function, aging, degeneration, dystrophy and regeneration of skeletal muscle, with an emphasis on understanding adult skeletal muscle, its maintenance, and its interactions with non-muscle cell types and regulatory modulators.
Main areas of interest include:
-differentiation of skeletal muscle-
atrophy and hypertrophy of skeletal muscle-
aging of skeletal muscle-
regeneration and degeneration of skeletal muscle-
biology of satellite and satellite-like cells-
dystrophic degeneration of skeletal muscle-
energy and glucose homeostasis in skeletal muscle-
non-dystrophic genetic diseases of skeletal muscle, such as Spinal Muscular Atrophy and myopathies-
maintenance of neuromuscular junctions-
roles of ryanodine receptors and calcium signaling in skeletal muscle-
roles of nuclear receptors in skeletal muscle-
roles of GPCRs and GPCR signaling in skeletal muscle-
other relevant aspects of skeletal muscle biology.
In addition, articles on translational clinical studies that address molecular and cellular mechanisms of skeletal muscle will be published. Case reports are also encouraged for submission.
Skeletal Muscle reflects the breadth of research on skeletal muscle and bridges gaps between diverse areas of science for example cardiac cell biology and neurobiology, which share common features with respect to cell differentiation, excitatory membranes, cell-cell communication, and maintenance. Suitable articles are model and mechanism-driven, and apply statistical principles where appropriate; purely descriptive studies are of lesser interest.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
