Katharina E Meijboom, Emma R Sutton, Eve McCallion, Emily McFall, Daniel Anthony, Benjamin Edwards, Sabrina Kubinski, Ines Tapken, Ines Bünermann, Gareth Hazell, Nina Ahlskog, Peter Claus, Kay E Davies, Rashmi Kothary, Matthew J A Wood, Melissa Bowerman
{"title":"Dysregulation of Tweak and Fn14 in skeletal muscle of spinal muscular atrophy mice.","authors":"Katharina E Meijboom, Emma R Sutton, Eve McCallion, Emily McFall, Daniel Anthony, Benjamin Edwards, Sabrina Kubinski, Ines Tapken, Ines Bünermann, Gareth Hazell, Nina Ahlskog, Peter Claus, Kay E Davies, Rashmi Kothary, Matthew J A Wood, Melissa Bowerman","doi":"10.1186/s13395-022-00301-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Spinal muscular atrophy (SMA) is a childhood neuromuscular disorder caused by depletion of the survival motor neuron (SMN) protein. SMA is characterized by the selective death of spinal cord motor neurons, leading to progressive muscle wasting. Loss of skeletal muscle in SMA is a combination of denervation-induced muscle atrophy and intrinsic muscle pathologies. Elucidation of the pathways involved is essential to identify the key molecules that contribute to and sustain muscle pathology. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)/TNF receptor superfamily member fibroblast growth factor-inducible 14 (Fn14) pathway has been shown to play a critical role in the regulation of denervation-induced muscle atrophy as well as muscle proliferation, differentiation, and metabolism in adults. However, it is not clear whether this pathway would be important in highly dynamic and developing muscle.</p><p><strong>Methods: </strong>We thus investigated the potential role of the TWEAK/Fn14 pathway in SMA muscle pathology, using the severe Taiwanese Smn<sup>-/-</sup>; SMN2 and the less severe Smn<sup>2B/-</sup> SMA mice, which undergo a progressive neuromuscular decline in the first three post-natal weeks. We also used experimental models of denervation and muscle injury in pre-weaned wild-type (WT) animals and siRNA-mediated knockdown in C2C12 muscle cells to conduct additional mechanistic investigations.</p><p><strong>Results: </strong>Here, we report significantly dysregulated expression of Tweak, Fn14, and previously proposed downstream effectors during disease progression in skeletal muscle of the two SMA mouse models. In addition, siRNA-mediated Smn knockdown in C2C12 myoblasts suggests a genetic interaction between Smn and the TWEAK/Fn14 pathway. Further analyses of SMA, Tweak<sup>-/-</sup>, and Fn14<sup>-/-</sup> mice revealed dysregulated myopathy, myogenesis, and glucose metabolism pathways as a common skeletal muscle feature, providing further evidence in support of a relationship between the TWEAK/Fn14 pathway and Smn. Finally, administration of the TWEAK/Fn14 agonist Fc-TWEAK improved disease phenotypes in the two SMA mouse models.</p><p><strong>Conclusions: </strong>Our study provides mechanistic insights into potential molecular players that contribute to muscle pathology in SMA and into likely differential responses of the TWEAK/Fn14 pathway in developing muscle.</p>","PeriodicalId":21747,"journal":{"name":"Skeletal Muscle","volume":" ","pages":"18"},"PeriodicalIF":4.4000,"publicationDate":"2022-07-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9331072/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Skeletal Muscle","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-022-00301-z","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Spinal muscular atrophy (SMA) is a childhood neuromuscular disorder caused by depletion of the survival motor neuron (SMN) protein. SMA is characterized by the selective death of spinal cord motor neurons, leading to progressive muscle wasting. Loss of skeletal muscle in SMA is a combination of denervation-induced muscle atrophy and intrinsic muscle pathologies. Elucidation of the pathways involved is essential to identify the key molecules that contribute to and sustain muscle pathology. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)/TNF receptor superfamily member fibroblast growth factor-inducible 14 (Fn14) pathway has been shown to play a critical role in the regulation of denervation-induced muscle atrophy as well as muscle proliferation, differentiation, and metabolism in adults. However, it is not clear whether this pathway would be important in highly dynamic and developing muscle.
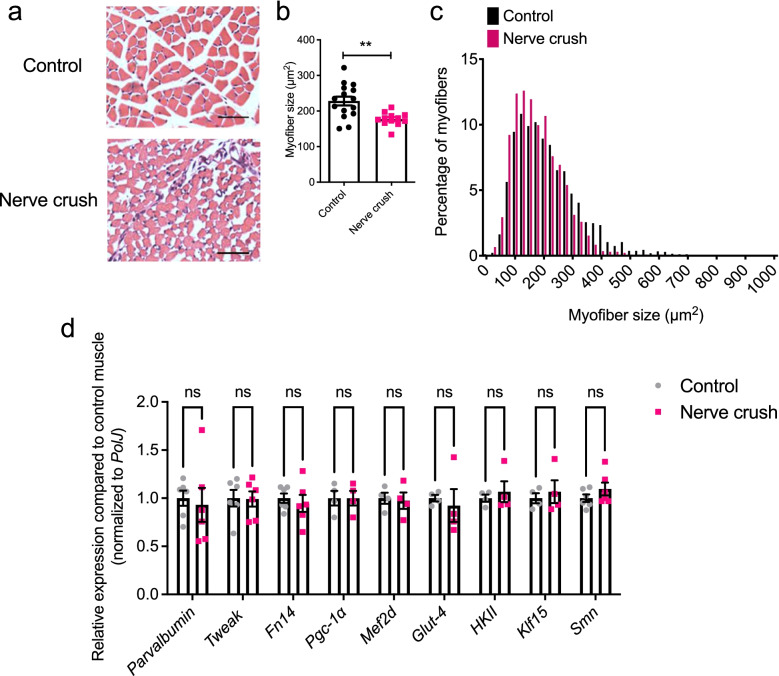
Methods: We thus investigated the potential role of the TWEAK/Fn14 pathway in SMA muscle pathology, using the severe Taiwanese Smn-/-; SMN2 and the less severe Smn2B/- SMA mice, which undergo a progressive neuromuscular decline in the first three post-natal weeks. We also used experimental models of denervation and muscle injury in pre-weaned wild-type (WT) animals and siRNA-mediated knockdown in C2C12 muscle cells to conduct additional mechanistic investigations.
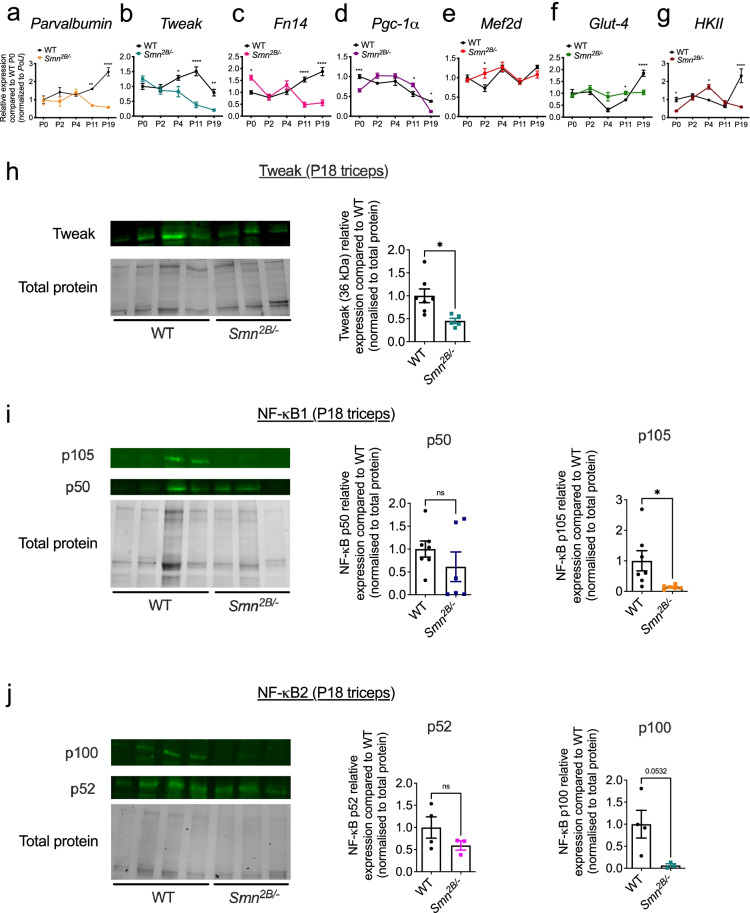
Results: Here, we report significantly dysregulated expression of Tweak, Fn14, and previously proposed downstream effectors during disease progression in skeletal muscle of the two SMA mouse models. In addition, siRNA-mediated Smn knockdown in C2C12 myoblasts suggests a genetic interaction between Smn and the TWEAK/Fn14 pathway. Further analyses of SMA, Tweak-/-, and Fn14-/- mice revealed dysregulated myopathy, myogenesis, and glucose metabolism pathways as a common skeletal muscle feature, providing further evidence in support of a relationship between the TWEAK/Fn14 pathway and Smn. Finally, administration of the TWEAK/Fn14 agonist Fc-TWEAK improved disease phenotypes in the two SMA mouse models.
Conclusions: Our study provides mechanistic insights into potential molecular players that contribute to muscle pathology in SMA and into likely differential responses of the TWEAK/Fn14 pathway in developing muscle.
期刊介绍:
The only open access journal in its field, Skeletal Muscle publishes novel, cutting-edge research and technological advancements that investigate the molecular mechanisms underlying the biology of skeletal muscle. Reflecting the breadth of research in this area, the journal welcomes manuscripts about the development, metabolism, the regulation of mass and function, aging, degeneration, dystrophy and regeneration of skeletal muscle, with an emphasis on understanding adult skeletal muscle, its maintenance, and its interactions with non-muscle cell types and regulatory modulators.
Main areas of interest include:
-differentiation of skeletal muscle-
atrophy and hypertrophy of skeletal muscle-
aging of skeletal muscle-
regeneration and degeneration of skeletal muscle-
biology of satellite and satellite-like cells-
dystrophic degeneration of skeletal muscle-
energy and glucose homeostasis in skeletal muscle-
non-dystrophic genetic diseases of skeletal muscle, such as Spinal Muscular Atrophy and myopathies-
maintenance of neuromuscular junctions-
roles of ryanodine receptors and calcium signaling in skeletal muscle-
roles of nuclear receptors in skeletal muscle-
roles of GPCRs and GPCR signaling in skeletal muscle-
other relevant aspects of skeletal muscle biology.
In addition, articles on translational clinical studies that address molecular and cellular mechanisms of skeletal muscle will be published. Case reports are also encouraged for submission.
Skeletal Muscle reflects the breadth of research on skeletal muscle and bridges gaps between diverse areas of science for example cardiac cell biology and neurobiology, which share common features with respect to cell differentiation, excitatory membranes, cell-cell communication, and maintenance. Suitable articles are model and mechanism-driven, and apply statistical principles where appropriate; purely descriptive studies are of lesser interest.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
