Tiffany C Taylor, Bianca M Coleman, Samyuktha P Arunkumar, Ipsita Dey, John T Dillon, Nicole O Ponde, Amanda C Poholek, Daniella M Schwartz, Mandy J McGeachy, Heather R Conti, Sarah L Gaffen
{"title":"IκBζ is an essential mediator of immunity to oropharyngeal candidiasis.","authors":"Tiffany C Taylor, Bianca M Coleman, Samyuktha P Arunkumar, Ipsita Dey, John T Dillon, Nicole O Ponde, Amanda C Poholek, Daniella M Schwartz, Mandy J McGeachy, Heather R Conti, Sarah L Gaffen","doi":"10.1016/j.chom.2023.08.016","DOIUrl":null,"url":null,"abstract":"<p><p>Fungal infections are a global threat; yet, there are no licensed vaccines to any fungal pathogens. Th17 cells mediate immunity to Candida albicans, particularly oropharyngeal candidiasis (OPC), but essential downstream mechanisms remain unclear. In the murine model of OPC, IκBζ (Nfkbiz, a non-canonical NF-κB transcription factor) was upregulated in an interleukin (IL)-17-dependent manner and was essential to prevent candidiasis. Deletion of Nfkbiz rendered mice highly susceptible to OPC. IκBζ was dispensable in hematopoietic cells and acted partially in the suprabasal oral epithelium to control OPC. One prominent IκBζ-dependent gene target was β-defensin 3 (BD3) (Defb3), an essential antimicrobial peptide. Human oral epithelial cells required IκBζ for IL-17-mediated induction of BD2 (DEFB4A, human ortholog of mouse Defb3) through binding to the DEFB4A promoter. Unexpectedly, IκBζ regulated the transcription factor Egr3, which was essential for C. albicans induction of BD2/DEFB4A. Accordingly, IκBζ and Egr3 comprise an antifungal signaling hub mediating mucosal defense against oral candidiasis.</p>","PeriodicalId":93926,"journal":{"name":"Cell host & microbe","volume":" ","pages":"1700-1713.e4"},"PeriodicalIF":18.7000,"publicationDate":"2023-10-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10591851/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell host & microbe","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.chom.2023.08.016","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/18 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
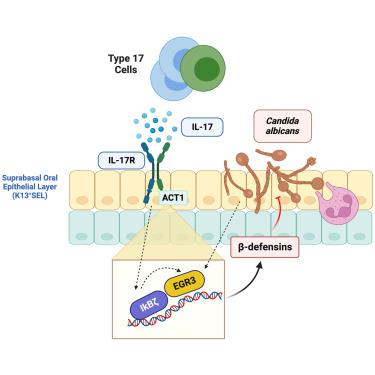
Fungal infections are a global threat; yet, there are no licensed vaccines to any fungal pathogens. Th17 cells mediate immunity to Candida albicans, particularly oropharyngeal candidiasis (OPC), but essential downstream mechanisms remain unclear. In the murine model of OPC, IκBζ (Nfkbiz, a non-canonical NF-κB transcription factor) was upregulated in an interleukin (IL)-17-dependent manner and was essential to prevent candidiasis. Deletion of Nfkbiz rendered mice highly susceptible to OPC. IκBζ was dispensable in hematopoietic cells and acted partially in the suprabasal oral epithelium to control OPC. One prominent IκBζ-dependent gene target was β-defensin 3 (BD3) (Defb3), an essential antimicrobial peptide. Human oral epithelial cells required IκBζ for IL-17-mediated induction of BD2 (DEFB4A, human ortholog of mouse Defb3) through binding to the DEFB4A promoter. Unexpectedly, IκBζ regulated the transcription factor Egr3, which was essential for C. albicans induction of BD2/DEFB4A. Accordingly, IκBζ and Egr3 comprise an antifungal signaling hub mediating mucosal defense against oral candidiasis.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
