{"title":"An in silico investigation of Kv2.1 potassium channel: Model building and inhibitors binding sites analysis.","authors":"Xiaoyu Wang, Xinyuan Zhang, Jie Zhou, Weiping Wang, Xiaoliang Wang, Bailing Xu","doi":"10.1002/minf.202300072","DOIUrl":null,"url":null,"abstract":"<p><p>Kv2.1 is widely expressed in brain, and inhibiting Kv2.1 is a potential strategy to prevent cell death and achieve neuroprotection in ischemic stroke. Herein, an in silico model of Kv2.1 tetramer structure was constructed by employing the AlphaFold-Multimer deep learning method to facilitate the rational discovery of Kv2.1 inhibitors. GaMD was utilized to create an ion transporting trajectory, which was analyzed with HMM to generate multiple representative receptor conformations. The binding site of RY785 and RY796(S) under the P-loop was defined with Fpocket program together with the competitive binding electrophysiology assay. The docking poses of the two inhibitors were predicted with the aid of the semi-empirical quantum mechanical calculation, and the IGMH results suggested that Met375, Thr376, and Thr377 of the P-helix and Ile405 of the S6 segment made significant contributions to the binding affinity. These results provided insights for rational molecular design to develop novel Kv2.1 inhibitors.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":" ","pages":"e202300072"},"PeriodicalIF":3.1000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202300072","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/7 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
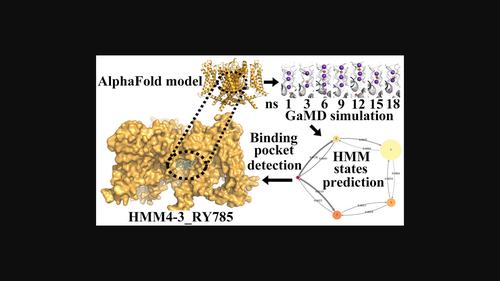
Kv2.1 is widely expressed in brain, and inhibiting Kv2.1 is a potential strategy to prevent cell death and achieve neuroprotection in ischemic stroke. Herein, an in silico model of Kv2.1 tetramer structure was constructed by employing the AlphaFold-Multimer deep learning method to facilitate the rational discovery of Kv2.1 inhibitors. GaMD was utilized to create an ion transporting trajectory, which was analyzed with HMM to generate multiple representative receptor conformations. The binding site of RY785 and RY796(S) under the P-loop was defined with Fpocket program together with the competitive binding electrophysiology assay. The docking poses of the two inhibitors were predicted with the aid of the semi-empirical quantum mechanical calculation, and the IGMH results suggested that Met375, Thr376, and Thr377 of the P-helix and Ile405 of the S6 segment made significant contributions to the binding affinity. These results provided insights for rational molecular design to develop novel Kv2.1 inhibitors.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
