Gabriel A Vignolle, Robert L Mach, Astrid R Mach-Aigner, Christian Zimmermann
{"title":"FunOrder 2.0 - a method for the fully automated curation of co-evolved genes in fungal biosynthetic gene clusters.","authors":"Gabriel A Vignolle, Robert L Mach, Astrid R Mach-Aigner, Christian Zimmermann","doi":"10.3389/ffunb.2022.1020623","DOIUrl":null,"url":null,"abstract":"<p><p>Coevolution is an important biological process that shapes interacting proteins - may it be physically interacting proteins or consecutive enzymes in a metabolic pathway, such as the biosynthetic pathways for secondary metabolites. Previously, we developed FunOrder, a semi-automated method for the detection of co-evolved genes, and demonstrated that FunOrder can be used to identify essential genes in biosynthetic gene clusters from different ascomycetes. A major drawback of this original method was the need for a manual assessment, which may create a user bias and prevents a high-throughput application. Here we present a fully automated version of this method termed FunOrder 2.0. In the improved version, we use several mathematical indices to determine the optimal number of clusters in the FunOrder output, and a subsequent k-means clustering based on the first three principal components of a principal component analysis of the FunOrder output to automatically detect co-evolved genes. Further, we replaced the BLAST tool with the DIAMOND tool as a prerequisite for using larger proteome databases. Potentially, FunOrder 2.0 may be used for the assessment of complete genomes, which has not been attempted yet. However, the introduced changes slightly decreased the sensitivity of this method, which is outweighed by enhanced overall speed and specificity.</p>","PeriodicalId":73084,"journal":{"name":"Frontiers in fungal biology","volume":"3 ","pages":"1020623"},"PeriodicalIF":2.1000,"publicationDate":"2022-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10512238/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in fungal biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/ffunb.2022.1020623","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MYCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
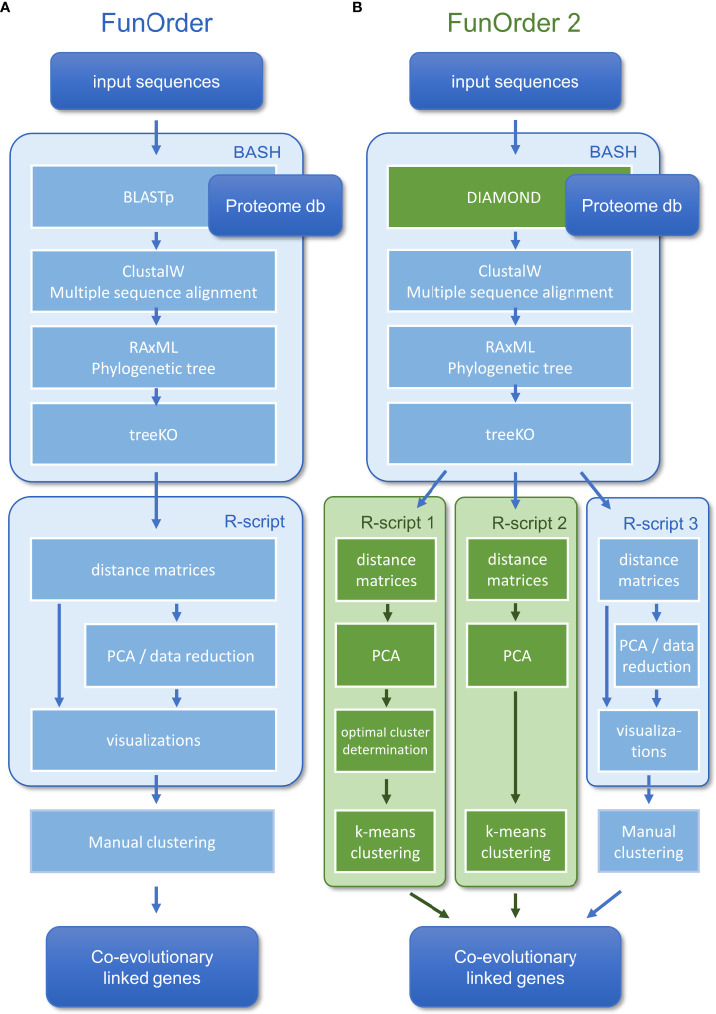
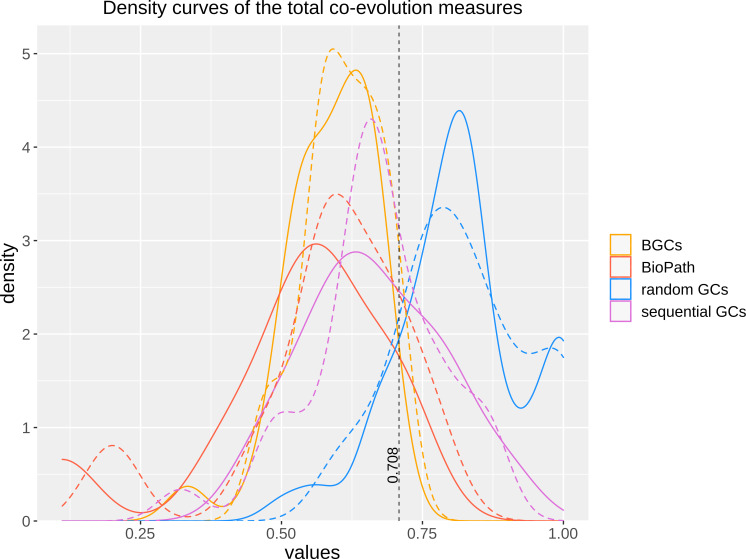
Coevolution is an important biological process that shapes interacting proteins - may it be physically interacting proteins or consecutive enzymes in a metabolic pathway, such as the biosynthetic pathways for secondary metabolites. Previously, we developed FunOrder, a semi-automated method for the detection of co-evolved genes, and demonstrated that FunOrder can be used to identify essential genes in biosynthetic gene clusters from different ascomycetes. A major drawback of this original method was the need for a manual assessment, which may create a user bias and prevents a high-throughput application. Here we present a fully automated version of this method termed FunOrder 2.0. In the improved version, we use several mathematical indices to determine the optimal number of clusters in the FunOrder output, and a subsequent k-means clustering based on the first three principal components of a principal component analysis of the FunOrder output to automatically detect co-evolved genes. Further, we replaced the BLAST tool with the DIAMOND tool as a prerequisite for using larger proteome databases. Potentially, FunOrder 2.0 may be used for the assessment of complete genomes, which has not been attempted yet. However, the introduced changes slightly decreased the sensitivity of this method, which is outweighed by enhanced overall speed and specificity.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
