Shangqian Xie, Karissa Isaacs, Gabrielle Becker, Brenda M Murdoch
{"title":"A computational framework for improving genetic variants identification from 5,061 sheep sequencing data.","authors":"Shangqian Xie, Karissa Isaacs, Gabrielle Becker, Brenda M Murdoch","doi":"10.1186/s40104-023-00923-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pan-genomics is a recently emerging strategy that can be utilized to provide a more comprehensive characterization of genetic variation. Joint calling is routinely used to combine identified variants across multiple related samples. However, the improvement of variants identification using the mutual support information from multiple samples remains quite limited for population-scale genotyping.</p><p><strong>Results: </strong>In this study, we developed a computational framework for joint calling genetic variants from 5,061 sheep by incorporating the sequencing error and optimizing mutual support information from multiple samples' data. The variants were accurately identified from multiple samples by using four steps: (1) Probabilities of variants from two widely used algorithms, GATK and Freebayes, were calculated by Poisson model incorporating base sequencing error potential; (2) The variants with high mapping quality or consistently identified from at least two samples by GATK and Freebayes were used to construct the raw high-confidence identification (rHID) variants database; (3) The high confidence variants identified in single sample were ordered by probability value and controlled by false discovery rate (FDR) using rHID database; (4) To avoid the elimination of potentially true variants from rHID database, the variants that failed FDR were reexamined to rescued potential true variants and ensured high accurate identification variants. The results indicated that the percent of concordant SNPs and Indels from Freebayes and GATK after our new method were significantly improved 12%-32% compared with raw variants and advantageously found low frequency variants of individual sheep involved several traits including nipples number (GPC5), scrapie pathology (PAPSS2), seasonal reproduction and litter size (GRM1), coat color (RAB27A), and lentivirus susceptibility (TMEM154).</p><p><strong>Conclusion: </strong>The new method used the computational strategy to reduce the number of false positives, and simultaneously improve the identification of genetic variants. This strategy did not incur any extra cost by using any additional samples or sequencing data information and advantageously identified rare variants which can be important for practical applications of animal breeding.</p>","PeriodicalId":64067,"journal":{"name":"Journal of Animal Science and Biotechnology","volume":"14 1","pages":"127"},"PeriodicalIF":6.5000,"publicationDate":"2023-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10544426/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Animal Science and Biotechnology","FirstCategoryId":"1089","ListUrlMain":"https://doi.org/10.1186/s40104-023-00923-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Pan-genomics is a recently emerging strategy that can be utilized to provide a more comprehensive characterization of genetic variation. Joint calling is routinely used to combine identified variants across multiple related samples. However, the improvement of variants identification using the mutual support information from multiple samples remains quite limited for population-scale genotyping.
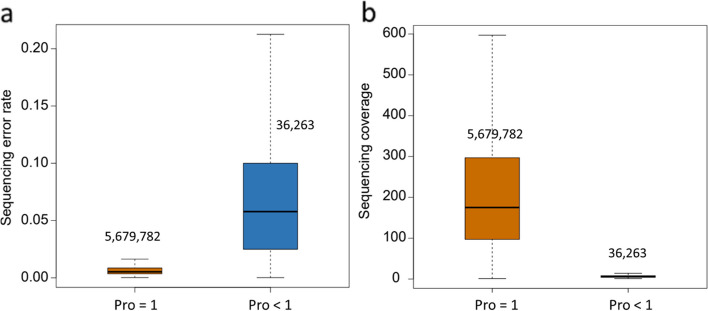
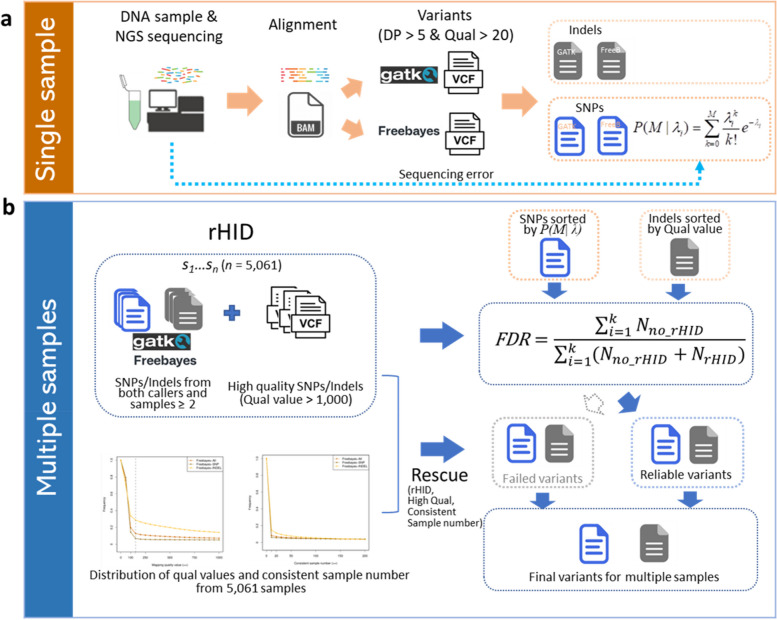
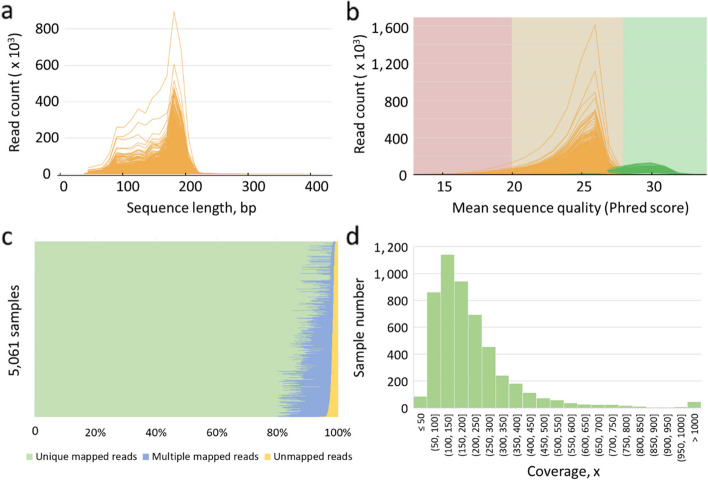
Results: In this study, we developed a computational framework for joint calling genetic variants from 5,061 sheep by incorporating the sequencing error and optimizing mutual support information from multiple samples' data. The variants were accurately identified from multiple samples by using four steps: (1) Probabilities of variants from two widely used algorithms, GATK and Freebayes, were calculated by Poisson model incorporating base sequencing error potential; (2) The variants with high mapping quality or consistently identified from at least two samples by GATK and Freebayes were used to construct the raw high-confidence identification (rHID) variants database; (3) The high confidence variants identified in single sample were ordered by probability value and controlled by false discovery rate (FDR) using rHID database; (4) To avoid the elimination of potentially true variants from rHID database, the variants that failed FDR were reexamined to rescued potential true variants and ensured high accurate identification variants. The results indicated that the percent of concordant SNPs and Indels from Freebayes and GATK after our new method were significantly improved 12%-32% compared with raw variants and advantageously found low frequency variants of individual sheep involved several traits including nipples number (GPC5), scrapie pathology (PAPSS2), seasonal reproduction and litter size (GRM1), coat color (RAB27A), and lentivirus susceptibility (TMEM154).
Conclusion: The new method used the computational strategy to reduce the number of false positives, and simultaneously improve the identification of genetic variants. This strategy did not incur any extra cost by using any additional samples or sequencing data information and advantageously identified rare variants which can be important for practical applications of animal breeding.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
