A Recurrent Nonsense Mutation in NECTIN4 Underlying Ectodermal Dysplasia-Syndactyly Syndrome with a Novel Phenotype in a Consanguineous Kashmiri Family.
Ghazanfar Ali, Sadia Sadia, Syeda Ain-Ul-Batool, Zahid Azeem, Naheed Bashir Awan, Syed Akif Raza Kazmi, Zia- Ur-Rehman, Zeeshan Anjum, Fazal- Ur-Rehman, Abdul Wali, Kafaitullah Khan, Nasib Zaman, Muhammad Ayub, Muhammad Sajid, Noor Hassan
{"title":"A Recurrent Nonsense Mutation in NECTIN4 Underlying Ectodermal Dysplasia-Syndactyly Syndrome with a Novel Phenotype in a Consanguineous Kashmiri Family.","authors":"Ghazanfar Ali, Sadia Sadia, Syeda Ain-Ul-Batool, Zahid Azeem, Naheed Bashir Awan, Syed Akif Raza Kazmi, Zia- Ur-Rehman, Zeeshan Anjum, Fazal- Ur-Rehman, Abdul Wali, Kafaitullah Khan, Nasib Zaman, Muhammad Ayub, Muhammad Sajid, Noor Hassan","doi":"10.1155/2023/9999660","DOIUrl":null,"url":null,"abstract":"<p><p>EDSS1, a syndrome characterized by ectodermal dysplasia-syndactyly, is inherited in an autosomal recessive manner due to mutations in the NECTIN4/PVRL4 gene. Clinical manifestations of the syndrome include defective nail plate, sparse to absent scalp and body hair, spaced teeth with enamel hypoplasia, and bilateral cutaneous syndactyly in the fingers and toes. Here, we report a consanguineous family of Kashmiri origin presenting features of EDSS1. Using whole exome sequencing, we found a recurrent nonsense mutation (NM_030916: c.181C > T, p.(Gln61 <i>∗</i>)) in the NECTIN4 gene. The variant segregated perfectly with the disorder within the family. The candidate variant was absent in 50 in-house exomes pertaining to other disorders from the same population. In addition to the previously reported clinical phenotype, an upper lip cleft was found in one of the affected members as a novel phenotype that is not reported by previous studies in EDSS1 patients. Therefore, the study presented here, which was conducted on the Kashmiri population, is the first to document a NECTIN4 mutation associated with the upper lip cleft as a novel phenotype. This finding broadens the molecular and phenotypic spectrum of EDSS1.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2023 ","pages":"9999660"},"PeriodicalIF":2.1000,"publicationDate":"2023-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10567209/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/2023/9999660","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
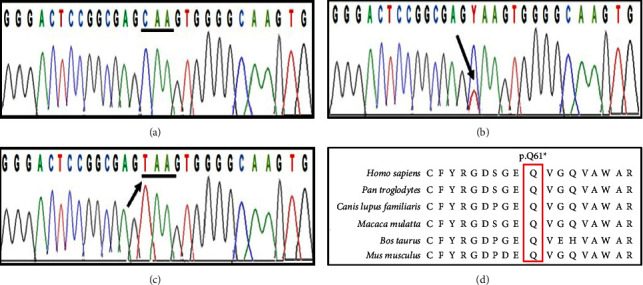
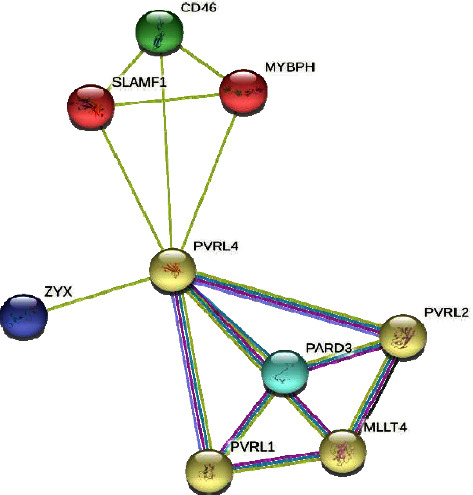
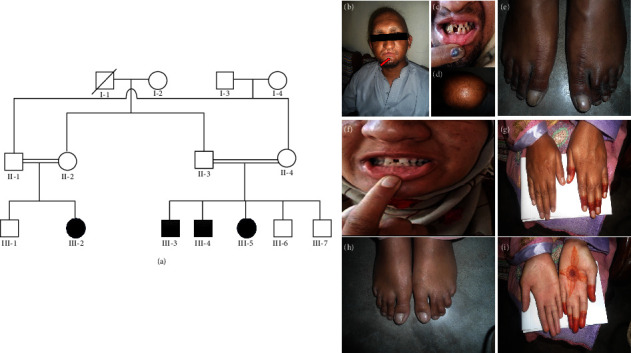
EDSS1, a syndrome characterized by ectodermal dysplasia-syndactyly, is inherited in an autosomal recessive manner due to mutations in the NECTIN4/PVRL4 gene. Clinical manifestations of the syndrome include defective nail plate, sparse to absent scalp and body hair, spaced teeth with enamel hypoplasia, and bilateral cutaneous syndactyly in the fingers and toes. Here, we report a consanguineous family of Kashmiri origin presenting features of EDSS1. Using whole exome sequencing, we found a recurrent nonsense mutation (NM_030916: c.181C > T, p.(Gln61 ∗)) in the NECTIN4 gene. The variant segregated perfectly with the disorder within the family. The candidate variant was absent in 50 in-house exomes pertaining to other disorders from the same population. In addition to the previously reported clinical phenotype, an upper lip cleft was found in one of the affected members as a novel phenotype that is not reported by previous studies in EDSS1 patients. Therefore, the study presented here, which was conducted on the Kashmiri population, is the first to document a NECTIN4 mutation associated with the upper lip cleft as a novel phenotype. This finding broadens the molecular and phenotypic spectrum of EDSS1.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
