Emma S Singer, Joshua Crowe, Mira Holliday, Julia C Isbister, Sean Lal, Natalie Nowak, Laura Yeates, Charlotte Burns, Sulekha Rajagopalan, Ivan Macciocca, Ingrid King, Julie Wacker, Jodie Ingles, Robert G Weintraub, Christopher Semsarian, Richard D Bagnall
{"title":"The burden of splice-disrupting variants in inherited heart disease and unexplained sudden cardiac death.","authors":"Emma S Singer, Joshua Crowe, Mira Holliday, Julia C Isbister, Sean Lal, Natalie Nowak, Laura Yeates, Charlotte Burns, Sulekha Rajagopalan, Ivan Macciocca, Ingrid King, Julie Wacker, Jodie Ingles, Robert G Weintraub, Christopher Semsarian, Richard D Bagnall","doi":"10.1038/s41525-023-00373-w","DOIUrl":null,"url":null,"abstract":"<p><p>There is an incomplete understanding of the burden of splice-disrupting variants in definitively associated inherited heart disease genes and whether these genes can amplify from blood RNA to support functional confirmation of splicing outcomes. We performed burden testing of rare splice-disrupting variants in people with inherited heart disease and sudden unexplained death compared to 125,748 population controls. ClinGen definitively disease-associated inherited heart disease genes were amplified using RNA extracted from fresh blood, derived cardiomyocytes, and myectomy tissue. Variants were functionally assessed and classified for pathogenicity. We found 88 in silico-predicted splice-disrupting variants in 128 out of 1242 (10.3%) unrelated participants. There was an excess burden of splice-disrupting variants in PKP2 (5.9%), FLNC (2.7%), TTN (2.8%), MYBPC3 (8.2%) and MYH7 (1.3%), in distinct cardiomyopathy subtypes, and KCNQ1 (3.6%) in long QT syndrome. Blood RNA supported the amplification of 21 out of 31 definitive disease-associated inherited heart disease genes. Our functional studies confirmed altered splicing in six variants. Eleven variants of uncertain significance were reclassified as likely pathogenic based on functional studies and six were used for cascade genetic testing in 12 family members. Our study highlights that splice-disrupting variants are a significant cause of inherited heart disease, and that analysis of blood RNA confirms splicing outcomes and supports variant pathogenicity classification.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"8 1","pages":"29"},"PeriodicalIF":4.8000,"publicationDate":"2023-10-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10567745/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-023-00373-w","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
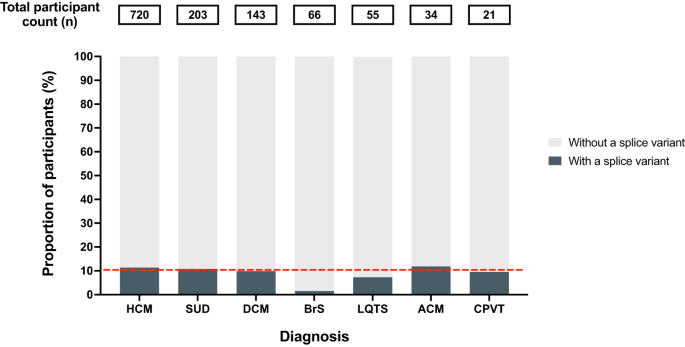
There is an incomplete understanding of the burden of splice-disrupting variants in definitively associated inherited heart disease genes and whether these genes can amplify from blood RNA to support functional confirmation of splicing outcomes. We performed burden testing of rare splice-disrupting variants in people with inherited heart disease and sudden unexplained death compared to 125,748 population controls. ClinGen definitively disease-associated inherited heart disease genes were amplified using RNA extracted from fresh blood, derived cardiomyocytes, and myectomy tissue. Variants were functionally assessed and classified for pathogenicity. We found 88 in silico-predicted splice-disrupting variants in 128 out of 1242 (10.3%) unrelated participants. There was an excess burden of splice-disrupting variants in PKP2 (5.9%), FLNC (2.7%), TTN (2.8%), MYBPC3 (8.2%) and MYH7 (1.3%), in distinct cardiomyopathy subtypes, and KCNQ1 (3.6%) in long QT syndrome. Blood RNA supported the amplification of 21 out of 31 definitive disease-associated inherited heart disease genes. Our functional studies confirmed altered splicing in six variants. Eleven variants of uncertain significance were reclassified as likely pathogenic based on functional studies and six were used for cascade genetic testing in 12 family members. Our study highlights that splice-disrupting variants are a significant cause of inherited heart disease, and that analysis of blood RNA confirms splicing outcomes and supports variant pathogenicity classification.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
