Sophie Baker, Joshua Pagotto, Timothy T. Duignan and Alister J. Page*,
{"title":"High-Throughput Aqueous Electrolyte Structure Prediction Using IonSolvR and Equivariant Graph Neural Network Potentials","authors":"Sophie Baker, Joshua Pagotto, Timothy T. Duignan and Alister J. Page*, ","doi":"10.1021/acs.jpclett.3c01783","DOIUrl":null,"url":null,"abstract":"<p >Neural network potentials have recently emerged as an efficient and accurate tool for accelerating <i>ab initio</i> molecular dynamics (AIMD) in order to simulate complex condensed phases such as electrolyte solutions. Their principal limitation, however, is their requirement for sufficiently large and accurate training sets, which are often composed of Kohn–Sham density functional theory (DFT) calculations. Here we examine the feasibility of using existing density functional tight-binding (DFTB) molecular dynamics trajectory data available in the IonSolvR database in order to accelerate the training of E(3)-equivariant graph neural network potentials. We show that the solvation structure of Na<sup>+</sup> and Cl<sup>–</sup> in aqueous NaCl solutions can be accurately reproduced with remarkably small amounts of data (i.e., 100 MD frames). We further show that these predictions can be systematically improved further via an embarrassingly parallel resampling approach.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"14 42","pages":"9508–9515"},"PeriodicalIF":4.6000,"publicationDate":"2023-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01783","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
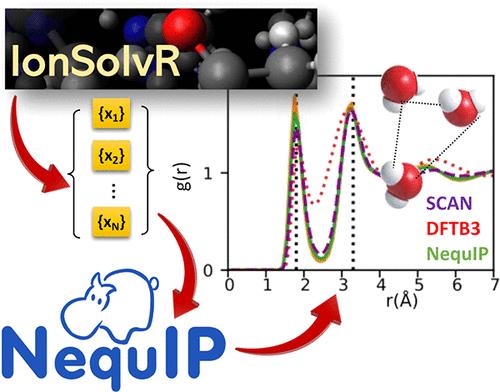
Neural network potentials have recently emerged as an efficient and accurate tool for accelerating ab initio molecular dynamics (AIMD) in order to simulate complex condensed phases such as electrolyte solutions. Their principal limitation, however, is their requirement for sufficiently large and accurate training sets, which are often composed of Kohn–Sham density functional theory (DFT) calculations. Here we examine the feasibility of using existing density functional tight-binding (DFTB) molecular dynamics trajectory data available in the IonSolvR database in order to accelerate the training of E(3)-equivariant graph neural network potentials. We show that the solvation structure of Na+ and Cl– in aqueous NaCl solutions can be accurately reproduced with remarkably small amounts of data (i.e., 100 MD frames). We further show that these predictions can be systematically improved further via an embarrassingly parallel resampling approach.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
