Research on the Reaction Mechanism of 2,4,6‐Trinitrotoluene Resource Utilization I:Formation of 2,4,6‐Trinitrobenzoic Acid in Acetic Acid with N,N',N″‐Trihydroxyisocyanuric Acid (THICA) Catalyst
{"title":"Research on the Reaction Mechanism of 2,4,6‐Trinitrotoluene Resource Utilization I:Formation of 2,4,6‐Trinitrobenzoic Acid in Acetic Acid with N,N',N″‐Trihydroxyisocyanuric Acid (THICA) Catalyst","authors":"Guan Zhang, Jin Li, Zongkuan Liu","doi":"10.1002/poc.4564","DOIUrl":null,"url":null,"abstract":"As an organic molecule catalyst, N,N',N\"‐trihydroxyisocyanuric acid can selectively catalyze the oxidation of the methyl group of waste 2,4,6‐trinitrotoluene to generate 2,4,6‐trinitrobenzoic acid. This reaction can avoid environmental pollution by inorganic heavy metal catalysts. In this study, four reaction stages of this catalytic reaction were designed and validated computationally at the M06‐2X‐D3ZERO/6‐311G(d,p) level using the acetic acid solvent model. These validations include transition state searches, intrinsic reaction coordinate calculations, reactant and product optimizations, and frequency calculations. The final reaction network of 23 transition states shows that after N,N',N\"‐trihydroxyisocyanuric acid activation and common reaction, the network bifurcates into two stages: alcohol to carboxylic acid and aldehyde to carboxylic acid. Although the former stage releases about 155 kcal/mol of Gibbs free energy, less than the 177 kcal/mol from the latter stage, the overall reaction equation shows that the pathway including former stage does not consume the catalytically active substance IM_T2, which saves the energy required for reactivation and is thus more favorable. Furthermore, the key transition states in the reaction network include bimolecular substitution reactions and proton hopping transfer reactions. Analyses of their interaction region indicators and intrinsic reaction coordinate results demonstrate strong selectivity. Additionally, the energy barriers and heat releases of the latter are twice and 1.3 times greater than those of the former, respectively. In summary, this study elucidated two competitive reaction pathways and identified the more energetically favorable and selective pathway, it provides useful insights for further optimization of industrial utilization of 2,4,6‐trinitrotoluene.","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2023-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1002/poc.4564","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
Abstract
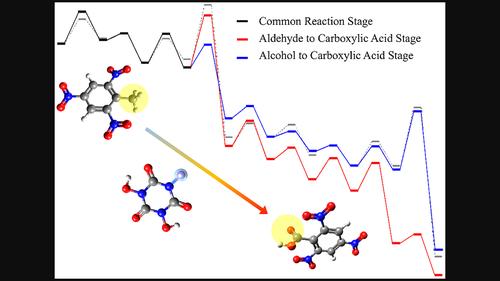
As an organic molecule catalyst, N,N',N"‐trihydroxyisocyanuric acid can selectively catalyze the oxidation of the methyl group of waste 2,4,6‐trinitrotoluene to generate 2,4,6‐trinitrobenzoic acid. This reaction can avoid environmental pollution by inorganic heavy metal catalysts. In this study, four reaction stages of this catalytic reaction were designed and validated computationally at the M06‐2X‐D3ZERO/6‐311G(d,p) level using the acetic acid solvent model. These validations include transition state searches, intrinsic reaction coordinate calculations, reactant and product optimizations, and frequency calculations. The final reaction network of 23 transition states shows that after N,N',N"‐trihydroxyisocyanuric acid activation and common reaction, the network bifurcates into two stages: alcohol to carboxylic acid and aldehyde to carboxylic acid. Although the former stage releases about 155 kcal/mol of Gibbs free energy, less than the 177 kcal/mol from the latter stage, the overall reaction equation shows that the pathway including former stage does not consume the catalytically active substance IM_T2, which saves the energy required for reactivation and is thus more favorable. Furthermore, the key transition states in the reaction network include bimolecular substitution reactions and proton hopping transfer reactions. Analyses of their interaction region indicators and intrinsic reaction coordinate results demonstrate strong selectivity. Additionally, the energy barriers and heat releases of the latter are twice and 1.3 times greater than those of the former, respectively. In summary, this study elucidated two competitive reaction pathways and identified the more energetically favorable and selective pathway, it provides useful insights for further optimization of industrial utilization of 2,4,6‐trinitrotoluene.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
