{"title":"N-heterocyclic carbene- and organic photoredox-catalysed meta-selective acylation of electron-rich arenes","authors":"Yamato Goto, Masaki Sano, Yuto Sumida, Hirohisa Ohmiya","doi":"10.1038/s44160-023-00378-4","DOIUrl":null,"url":null,"abstract":"meta-Selective functionalization of electron-rich arenes provides a complementary route to that of traditional organic synthesis. In classical electrophilic aromatic substitution reactions of electron-donating group-pendant arenes, C–H functionalization occurs at the ortho- or para-positions. There have been numerous efforts to overcome this selectivity, and various synthetic methods have been developed, typically using transition metal catalysis. Here we report a combined N-heterocyclic carbene- and organic photoredox-catalysed method for meta-selective acylation of electron-rich arenes, using acyl imidazoles as acylating reagents. This approach proceeds without directing groups or steric factors required in transition metal-catalysed processes, resulting in the opposite regioselectivity to conventional approaches such as Friedel–Crafts acylation. Mechanistic studies reveal the process involves a sequence of single-electron oxidation of an electron-rich arene followed by the radical–radical coupling between a ketyl radical and an arene radical cation. meta-Selective acylation of arenes typically requires directing groups or steric hindrance. Now, a combined N-heterocyclic carbene and organic photoredox catalysed of electron-rich arenes, using acyl imidazoles as acylating agents, is reported. Mechanistic studies reveal the process proceeds through single-electron oxidation and radical–radical coupling steps.","PeriodicalId":74251,"journal":{"name":"Nature synthesis","volume":"2 11","pages":"1037-1045"},"PeriodicalIF":20.0000,"publicationDate":"2023-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature synthesis","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s44160-023-00378-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"0","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 3
Abstract
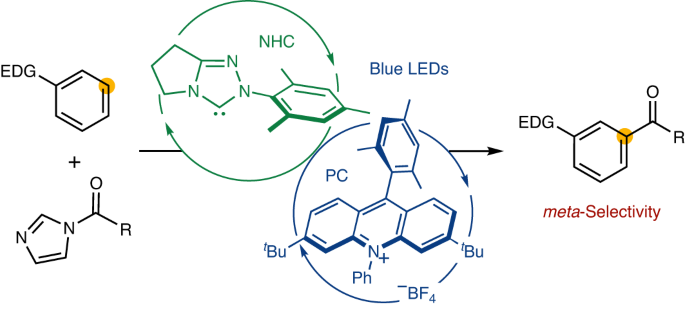
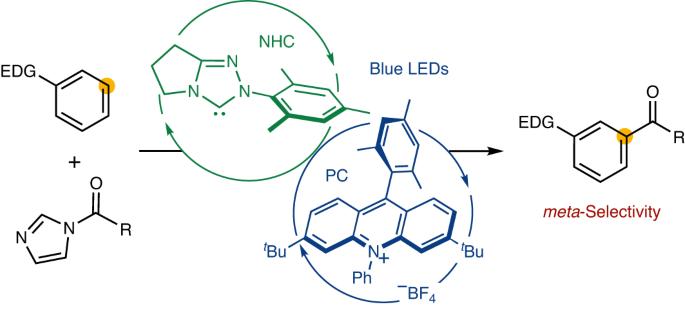
meta-Selective functionalization of electron-rich arenes provides a complementary route to that of traditional organic synthesis. In classical electrophilic aromatic substitution reactions of electron-donating group-pendant arenes, C–H functionalization occurs at the ortho- or para-positions. There have been numerous efforts to overcome this selectivity, and various synthetic methods have been developed, typically using transition metal catalysis. Here we report a combined N-heterocyclic carbene- and organic photoredox-catalysed method for meta-selective acylation of electron-rich arenes, using acyl imidazoles as acylating reagents. This approach proceeds without directing groups or steric factors required in transition metal-catalysed processes, resulting in the opposite regioselectivity to conventional approaches such as Friedel–Crafts acylation. Mechanistic studies reveal the process involves a sequence of single-electron oxidation of an electron-rich arene followed by the radical–radical coupling between a ketyl radical and an arene radical cation. meta-Selective acylation of arenes typically requires directing groups or steric hindrance. Now, a combined N-heterocyclic carbene and organic photoredox catalysed of electron-rich arenes, using acyl imidazoles as acylating agents, is reported. Mechanistic studies reveal the process proceeds through single-electron oxidation and radical–radical coupling steps.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
