{"title":"DFT investigations on the mechanisms and kinetics for the DMS + O3 reaction","authors":"Shuangjun Wang, Hui Zhao, Dong Chen, Chenggang Lu, Yizhen Tang","doi":"10.1002/poc.4558","DOIUrl":null,"url":null,"abstract":"<p>The potential energy surface (PES) for the reaction of ozone with dimethyl sulfide (DMS) was calculated at the CCSD(T)/6-311++G(3df,2pd)//M06-2X/6-311++G(d,p) levels of theory. Result shows that on the singlet PES the addition–elimination mechanism is dominant, and H-abstraction mechanism is less competitive. The major channel starts from the addition of ozone and DMS leading to a weak intermediate IM1, which decomposes subsequently to DMSO and <sup>1</sup>O<sub>2</sub> via a barrier around 38.8 kJ/mol. With a barrier of 64.0 kJ/mol, the formation of HO<sub>3</sub> + CH<sub>3</sub>SCH<sub>2</sub> via H-abstraction mechanism is subdominant. Besides, DMSO + <sup>1</sup>O<sub>2</sub> can take place further reactions to produce several products. The substitution mechanism was located on the triplet PES, however, with a rather high barrier it is negligible. Furthermore, the rate constants for the two channels leading to DMSO + <sup>1</sup>O<sub>2</sub> and HO<sub>3</sub> + CH<sub>3</sub>SCH<sub>2</sub> were calculated from 200 to 1000 K. The total rate constant is 1.13 × 10<sup>-20</sup> cm<sup>3</sup>·molecule<sup>-1</sup>·s<sup>-1</sup> at 298 K and 1 atm, in good agreement with previous experimental data. The overall rate constants are positive temperature dependent in the whole temperature range.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"36 11","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2023-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4558","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
Abstract
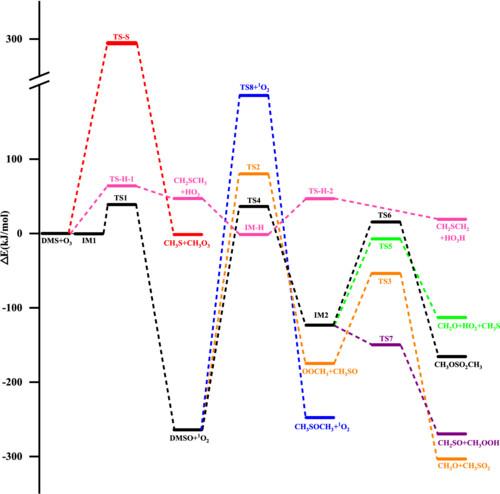
The potential energy surface (PES) for the reaction of ozone with dimethyl sulfide (DMS) was calculated at the CCSD(T)/6-311++G(3df,2pd)//M06-2X/6-311++G(d,p) levels of theory. Result shows that on the singlet PES the addition–elimination mechanism is dominant, and H-abstraction mechanism is less competitive. The major channel starts from the addition of ozone and DMS leading to a weak intermediate IM1, which decomposes subsequently to DMSO and 1O2 via a barrier around 38.8 kJ/mol. With a barrier of 64.0 kJ/mol, the formation of HO3 + CH3SCH2 via H-abstraction mechanism is subdominant. Besides, DMSO + 1O2 can take place further reactions to produce several products. The substitution mechanism was located on the triplet PES, however, with a rather high barrier it is negligible. Furthermore, the rate constants for the two channels leading to DMSO + 1O2 and HO3 + CH3SCH2 were calculated from 200 to 1000 K. The total rate constant is 1.13 × 10-20 cm3·molecule-1·s-1 at 298 K and 1 atm, in good agreement with previous experimental data. The overall rate constants are positive temperature dependent in the whole temperature range.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
