Mathew Chow, Tao E. Li and Sharon Hammes-Schiffer*,
{"title":"Nuclear–Electronic Orbital Quantum Mechanical/Molecular Mechanical Real-Time Dynamics","authors":"Mathew Chow, Tao E. Li and Sharon Hammes-Schiffer*, ","doi":"10.1021/acs.jpclett.3c02275","DOIUrl":null,"url":null,"abstract":"<p >Simulating the nuclear–electronic quantum dynamics of large-scale molecular systems in the condensed phase is key for studying biologically and chemically important processes such as proton transfer and proton-coupled electron transfer reactions. Herein, the real-time nuclear–electronic orbital time-dependent density functional theory (RT-NEO-TDDFT) approach is combined with a hybrid quantum mechanical/molecular mechanical (QM/MM) strategy to enable the accurate description of coupled nuclear–electronic quantum dynamics in the presence of heterogeneous environments such as solvent or proteins. The densities of the electrons and quantum protons are propagated in real time, while the other nuclei are propagated classically on the instantaneous electron–proton vibronic surface. This approach is applied to phenol bound to lysozyme, intramolecular proton transfer in malonaldehyde, and nonequilibrium excited-state intramolecular proton transfer in <i>o</i>-hydroxybenzaldehyde. These examples illustrate that the RT-NEO-TDDFT framework, coupled with an atomistic representation of the environment, allows the simulation of condensed-phase systems that exhibit significant nuclear quantum effects.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"14 43","pages":"9556–9562"},"PeriodicalIF":4.6000,"publicationDate":"2023-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.3c02275","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
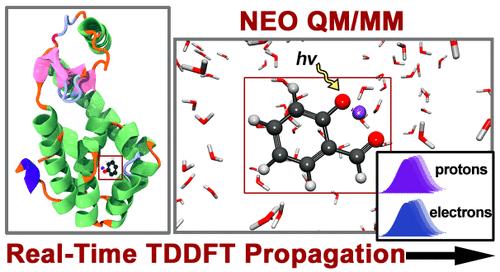
Simulating the nuclear–electronic quantum dynamics of large-scale molecular systems in the condensed phase is key for studying biologically and chemically important processes such as proton transfer and proton-coupled electron transfer reactions. Herein, the real-time nuclear–electronic orbital time-dependent density functional theory (RT-NEO-TDDFT) approach is combined with a hybrid quantum mechanical/molecular mechanical (QM/MM) strategy to enable the accurate description of coupled nuclear–electronic quantum dynamics in the presence of heterogeneous environments such as solvent or proteins. The densities of the electrons and quantum protons are propagated in real time, while the other nuclei are propagated classically on the instantaneous electron–proton vibronic surface. This approach is applied to phenol bound to lysozyme, intramolecular proton transfer in malonaldehyde, and nonequilibrium excited-state intramolecular proton transfer in o-hydroxybenzaldehyde. These examples illustrate that the RT-NEO-TDDFT framework, coupled with an atomistic representation of the environment, allows the simulation of condensed-phase systems that exhibit significant nuclear quantum effects.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
