Mira Seidel , Sandeep Rajkumar , Christina Steffke , Vivien Noeth , Shreya Agarwal , Kevin Roger , Joanna Lipecka , Albert Ludolph , Chiara Ida Guerrera , Tobias Boeckers , Alberto Catanese
{"title":"Propranolol reduces the accumulation of cytotoxic aggregates in C9orf72-ALS/FTD in vitro models","authors":"Mira Seidel , Sandeep Rajkumar , Christina Steffke , Vivien Noeth , Shreya Agarwal , Kevin Roger , Joanna Lipecka , Albert Ludolph , Chiara Ida Guerrera , Tobias Boeckers , Alberto Catanese","doi":"10.1016/j.crneur.2023.100105","DOIUrl":null,"url":null,"abstract":"<div><p>Mutations in the <em>C9orf72</em> gene are the most common cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The pathogenetic mechanisms linked to this gene are a direct consequence of an aberrant intronic expansion of a GGGGCC hexanucleotide located between the 1a and 1b non-coding exons, which can be transcribed to form cytotoxic RNA foci or even translated into aggregation-prone dipeptide repeat proteins. Importantly, the abnormal length of these repeats affects also the expression levels of C9orf72 itself, which suggests haploinsufficiency as additional pathomechanism. Thus, it appears that both toxic gain of function and loss of function are distinct but still coexistent features contributing to the insurgence of the disease in case of C9orf72 mutations. In this study, we aimed at identifying a strategy to address both aspects of the C9orf72-related pathobiochemistry and provide proof-of-principle information for a better understanding of the mechanisms leading to neuronal loss. By using primary neurons overexpressing toxic poly(GA), the most abundant protein product of the GGGGCC repeats, we found that the antiarrhythmic drug propranolol could efficiently reduce the accumulation of aberrant aggregates and increase the survival of C9orf72-related cultures. Interestingly, the improved catabolism appeared to not depend on major degradative pathways such as autophagy and the proteasome. By analyzing the proteome of poly(GA)-expressing neurons after exposure to propranolol, we found that the drug increased lysosomal degradation through a mechanism directly involving C9orf72 protein, whose levels were increased after treatment. Further confirmation of the beneficial effect of the beta blocker on aggregates' accumulation and survival of hiPSC-derived C9orf72-mutant motoneurons strengthened the finding that addressing both facets of C9orf72 pathology might represent a valid strategy for the treatment of these ALS/FTD cases.</p></div>","PeriodicalId":72752,"journal":{"name":"Current research in neurobiology","volume":"5 ","pages":"Article 100105"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current research in neurobiology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2665945X23000335","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
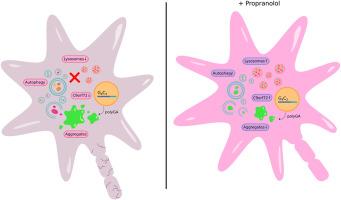
Mutations in the C9orf72 gene are the most common cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The pathogenetic mechanisms linked to this gene are a direct consequence of an aberrant intronic expansion of a GGGGCC hexanucleotide located between the 1a and 1b non-coding exons, which can be transcribed to form cytotoxic RNA foci or even translated into aggregation-prone dipeptide repeat proteins. Importantly, the abnormal length of these repeats affects also the expression levels of C9orf72 itself, which suggests haploinsufficiency as additional pathomechanism. Thus, it appears that both toxic gain of function and loss of function are distinct but still coexistent features contributing to the insurgence of the disease in case of C9orf72 mutations. In this study, we aimed at identifying a strategy to address both aspects of the C9orf72-related pathobiochemistry and provide proof-of-principle information for a better understanding of the mechanisms leading to neuronal loss. By using primary neurons overexpressing toxic poly(GA), the most abundant protein product of the GGGGCC repeats, we found that the antiarrhythmic drug propranolol could efficiently reduce the accumulation of aberrant aggregates and increase the survival of C9orf72-related cultures. Interestingly, the improved catabolism appeared to not depend on major degradative pathways such as autophagy and the proteasome. By analyzing the proteome of poly(GA)-expressing neurons after exposure to propranolol, we found that the drug increased lysosomal degradation through a mechanism directly involving C9orf72 protein, whose levels were increased after treatment. Further confirmation of the beneficial effect of the beta blocker on aggregates' accumulation and survival of hiPSC-derived C9orf72-mutant motoneurons strengthened the finding that addressing both facets of C9orf72 pathology might represent a valid strategy for the treatment of these ALS/FTD cases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
