Sadra Kashef Ol Gheta, Anne Bonin, Thomas Gerlach, Andreas H. Göller
{"title":"Predicting absolute aqueous solubility by applying a machine learning model for an artificially liquid-state as proxy for the solid-state","authors":"Sadra Kashef Ol Gheta, Anne Bonin, Thomas Gerlach, Andreas H. Göller","doi":"10.1007/s10822-023-00538-w","DOIUrl":null,"url":null,"abstract":"<div><p>In this study, we use machine learning algorithms with QM-derived COSMO-RS descriptors, along with Morgan fingerprints, to predict the absolute solubility of drug-like compounds. The QM-derived descriptors account for the molecular properties of the solute, i.e., the solute–solute interactions in an artificial-liquid-state (super-cooled liquid), and the solute–solvent interactions in solution. We employ two main approaches to predict solubility: (i) a hypothetical pathway that involves melting the solute at room temperature T = T¯ (<span>\\({\\Delta }_{fus}{G}_{A}^{\\ominus }\\)</span>) and mixing the artificially liquid solute into the solvent (<span>\\({\\Delta }_{m}{G}_{\\left(A:B\\right)}^{\\ominus }\\)</span>). In this approach <span>\\({\\Delta }_{fus}{G}_{A}^{\\ominus }\\)</span> is predicted using machine learning models, and the <span>\\({\\Delta }_{m}{G}_{\\left(A:B\\right)}^{\\ominus }\\)</span> is obtained from COSMO-RS calculations; (ii) direct solubility prediction using machine learning algorithms. The models were trained on a large number of Bayer in-house compounds for which water solubility data is available at physiological pH of 6.5 and ambient temperature. We also evaluated our models using external datasets from a solubility challenge. Our models present great improvements compared to the absolute solubility prediction with the QSAR model for the artificial liquid state as implemented in the COSMO<i>therm</i> software, for both in-house and external datasets. We are furthermore able to demonstrate the superiority of QM-derived descriptors compared to cheminformatics descriptors. We finally present low-cost alternative models using fragment-based COSMO<i>quick</i> calculations with only marginal reduction in the quality of predicted solubility.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 12","pages":"765 - 789"},"PeriodicalIF":3.1000,"publicationDate":"2023-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00538-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
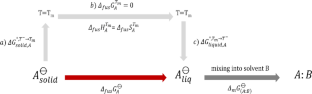
In this study, we use machine learning algorithms with QM-derived COSMO-RS descriptors, along with Morgan fingerprints, to predict the absolute solubility of drug-like compounds. The QM-derived descriptors account for the molecular properties of the solute, i.e., the solute–solute interactions in an artificial-liquid-state (super-cooled liquid), and the solute–solvent interactions in solution. We employ two main approaches to predict solubility: (i) a hypothetical pathway that involves melting the solute at room temperature T = T¯ (\({\Delta }_{fus}{G}_{A}^{\ominus }\)) and mixing the artificially liquid solute into the solvent (\({\Delta }_{m}{G}_{\left(A:B\right)}^{\ominus }\)). In this approach \({\Delta }_{fus}{G}_{A}^{\ominus }\) is predicted using machine learning models, and the \({\Delta }_{m}{G}_{\left(A:B\right)}^{\ominus }\) is obtained from COSMO-RS calculations; (ii) direct solubility prediction using machine learning algorithms. The models were trained on a large number of Bayer in-house compounds for which water solubility data is available at physiological pH of 6.5 and ambient temperature. We also evaluated our models using external datasets from a solubility challenge. Our models present great improvements compared to the absolute solubility prediction with the QSAR model for the artificial liquid state as implemented in the COSMOtherm software, for both in-house and external datasets. We are furthermore able to demonstrate the superiority of QM-derived descriptors compared to cheminformatics descriptors. We finally present low-cost alternative models using fragment-based COSMOquick calculations with only marginal reduction in the quality of predicted solubility.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
