{"title":"Electronic structures and properties of lead-free cesium- or rubidium-based perovskite halide compounds by first-principles calculations","authors":"Riku Okumura, Takeo Oku, Atsushi Suzuki","doi":"10.1016/j.nwnano.2023.100020","DOIUrl":null,"url":null,"abstract":"<div><p>Perovskite halide compounds are expected to provide various applications such as solar cells and light-emitting diodes. In the present work, structure models of ABX<sub>3</sub> (A = Rb, or Cs, B = Sn, Sr, or Ca, X = Cl, Br, or I) perovskite crystals were constructed, and the electronic structures and properties were analyzed by the first-principles calculations. It was found that halogen substitutions affected the energy gaps, and carrier mobilities of these perovskite crystals, and that the Sn-based perovskite crystals were predicted to have relatively low total energies and excellent carrier mobility. It was also found that the calculated total energies in this study are closely related with the tolerance factors, and the total energies decreased as the tolerance factors approach 1.</p></div>","PeriodicalId":100942,"journal":{"name":"Nano Trends","volume":"4 ","pages":"Article 100020"},"PeriodicalIF":0.0000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nano Trends","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2666978123000181","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/16 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
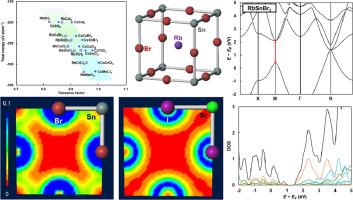
Perovskite halide compounds are expected to provide various applications such as solar cells and light-emitting diodes. In the present work, structure models of ABX3 (A = Rb, or Cs, B = Sn, Sr, or Ca, X = Cl, Br, or I) perovskite crystals were constructed, and the electronic structures and properties were analyzed by the first-principles calculations. It was found that halogen substitutions affected the energy gaps, and carrier mobilities of these perovskite crystals, and that the Sn-based perovskite crystals were predicted to have relatively low total energies and excellent carrier mobility. It was also found that the calculated total energies in this study are closely related with the tolerance factors, and the total energies decreased as the tolerance factors approach 1.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
