Clinical relevance of short-chain acyl-CoA dehydrogenase (SCAD) deficiency: Exploring the role of new variants including the first SCAD-disease-causing allele carrying a synonymous mutation
Rodolfo Tonin , Anna Caciotti , Silvia Funghini , Elisabetta Pasquini , Sean D. Mooney , Binghuang Cai , Elena Proncopio , Maria Alice Donati , Federico Baronio , Ilaria Bettocchi , Alessandra Cassio , Giacomo Biasucci , Andrea Bordugo , Giancarlo la Marca , Renzo Guerrini , Amelia Morrone
{"title":"Clinical relevance of short-chain acyl-CoA dehydrogenase (SCAD) deficiency: Exploring the role of new variants including the first SCAD-disease-causing allele carrying a synonymous mutation","authors":"Rodolfo Tonin , Anna Caciotti , Silvia Funghini , Elisabetta Pasquini , Sean D. Mooney , Binghuang Cai , Elena Proncopio , Maria Alice Donati , Federico Baronio , Ilaria Bettocchi , Alessandra Cassio , Giacomo Biasucci , Andrea Bordugo , Giancarlo la Marca , Renzo Guerrini , Amelia Morrone","doi":"10.1016/j.bbacli.2016.03.004","DOIUrl":null,"url":null,"abstract":"<div><p>Short-chain acyl-coA dehydrogenase deficiency (SCADD) is an autosomal recessive inborn error of mitochondrial fatty acid oxidation caused by <em>ACADS</em> gene alterations. SCADD is a heterogeneous condition, sometimes considered to be solely a biochemical condition given that it has been associated with variable clinical phenotypes ranging from no symptoms or signs to metabolic decompensation occurring early in life.</p><p>A reason for this variability is due to SCAD alterations, such as the common p.Gly209Ser, that confer a disease susceptibility state but require a complex multifactorial/polygenic condition to manifest clinically.</p><p>Our study focuses on 12 SCADD patients carrying 11 new <em>ACADS</em> variants, with the purpose of defining genotype–phenotype correlations based on clinical data, metabolite evaluation, molecular analyses, and <em>in silico</em> functional analyses.</p><p>Interestingly, we identified a synonymous variant, c.765G<!--> <!-->><!--> <!-->T (p.Gly255Gly) that influences <em>ACADS</em> mRNA splicing accuracy. mRNA characterisation demonstrated that this variant leads to an aberrant splicing product, harbouring a premature stop codon.</p><p>Molecular analysis and <em>in silico</em> tools are able to characterise <em>ACADS</em> variants, identifying the severe mutations and consequently indicating which patients could benefit from a long term follow- up. We also emphasise that synonymous mutations can be relevant features and potentially associated with SCADD.</p></div>","PeriodicalId":72344,"journal":{"name":"BBA clinical","volume":"5 ","pages":"Pages 114-119"},"PeriodicalIF":0.0000,"publicationDate":"2016-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.bbacli.2016.03.004","citationCount":"22","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BBA clinical","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2214647416300083","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 22
Abstract
Short-chain acyl-coA dehydrogenase deficiency (SCADD) is an autosomal recessive inborn error of mitochondrial fatty acid oxidation caused by ACADS gene alterations. SCADD is a heterogeneous condition, sometimes considered to be solely a biochemical condition given that it has been associated with variable clinical phenotypes ranging from no symptoms or signs to metabolic decompensation occurring early in life.
A reason for this variability is due to SCAD alterations, such as the common p.Gly209Ser, that confer a disease susceptibility state but require a complex multifactorial/polygenic condition to manifest clinically.
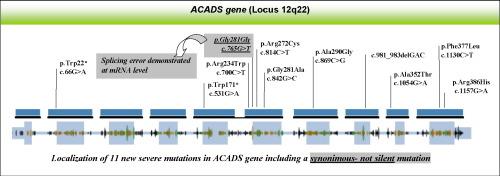
Our study focuses on 12 SCADD patients carrying 11 new ACADS variants, with the purpose of defining genotype–phenotype correlations based on clinical data, metabolite evaluation, molecular analyses, and in silico functional analyses.
Interestingly, we identified a synonymous variant, c.765G > T (p.Gly255Gly) that influences ACADS mRNA splicing accuracy. mRNA characterisation demonstrated that this variant leads to an aberrant splicing product, harbouring a premature stop codon.
Molecular analysis and in silico tools are able to characterise ACADS variants, identifying the severe mutations and consequently indicating which patients could benefit from a long term follow- up. We also emphasise that synonymous mutations can be relevant features and potentially associated with SCADD.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
