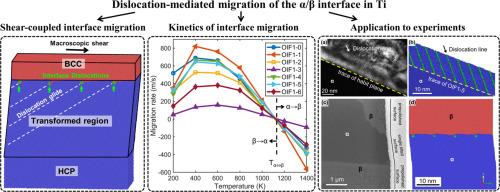
Interphase boundaries are essential in the deformation and phase transformations in titanium (Ti) alloys. While static structures of semicoherent α/β interfaces in various Ti alloys have been carefully examined, their migration behavior at atomic scales is far less clear. In this study, we employed molecular dynamics simulations to investigate the migration of the semicoherent α/β interface in pure Ti. The interface migration behavior shows a shear-coupled feature with the interface dislocation glide and a macroscopic shear. The simulation reveals that both the glide direction of the dislocations with respect to the interface and the dislocation spacing strongly influence the migration rate, and the low-index glide plane of the interface dislocation plays a minor role. The dependence of interface mobility on temperatures confirms the critical role of thermal activation during the interface migration, especially for activating the interface dislocation glide. Furthermore, the shear-coupled interface migration driven by element partition is simulated using a newly developed Ti-Mo potential, consistent with the displacive-diffusional features previously observed in the surface precipitates. The simulated interface migration mode is validated by comparing it with the crystallography features of surface precipitates in a Ti-Cr alloy. The interface energy and mobility obtained from simulations further explain why the distinctive crystallographic features of the surface precipitates observed experimentally are favored over other candidate interfaces. The present study has explored an approach for systematically examining thermodynamic and kinetic factors governing the development of phase transformation crystallography at different temperatures and chemical driving forces.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
