{"title":"Characterization of transcriptional enhancers in the chicken genome using CRISPR-mediated activation.","authors":"Jeong Hoon Han, Hong Jo Lee, Tae Hyun Kim","doi":"10.3389/fgeed.2023.1269115","DOIUrl":null,"url":null,"abstract":"<p><p>DNA regulatory elements intricately control when, where, and how genes are activated. Therefore, understanding the function of these elements could unveil the complexity of the genetic regulation network. Genome-wide significant variants are predominantly found in non-coding regions of DNA, so comprehending the predicted functional regulatory elements is crucial for understanding the biological context of these genomic markers, which can be incorporated into breeding programs. The emergence of CRISPR technology has provided a powerful tool for studying non-coding regulatory elements in genomes. In this study, we leveraged epigenetic data from the Functional Annotation of Animal Genomes project to identify promoter and putative enhancer regions associated with three genes (<i>HBBA, IRF7</i>, and <i>PPARG</i>) in the chicken genome. To identify the enhancer regions, we designed guide RNAs targeting the promoter and candidate enhancer regions and utilized CRISPR activation (CRISPRa) with dCas9-p300 and dCas9-VPR as transcriptional activators in chicken DF-1 cells. By comparing the expression levels of target genes between the promoter activation and the co-activation of the promoter and putative enhancers, we were able to identify functional enhancers that exhibited augmented upregulation. In conclusion, our findings demonstrate the remarkable efficiency of CRISPRa in precisely manipulating the expression of endogenous genes by targeting regulatory elements in the chicken genome, highlighting its potential for functional validation of non-coding regions.</p>","PeriodicalId":73086,"journal":{"name":"Frontiers in genome editing","volume":"5 ","pages":"1269115"},"PeriodicalIF":4.4000,"publicationDate":"2023-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10634339/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in genome editing","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fgeed.2023.1269115","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
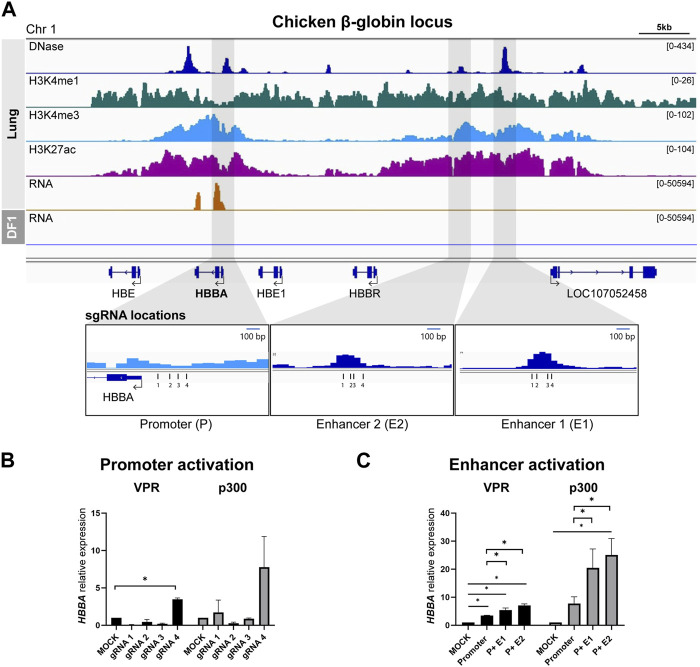
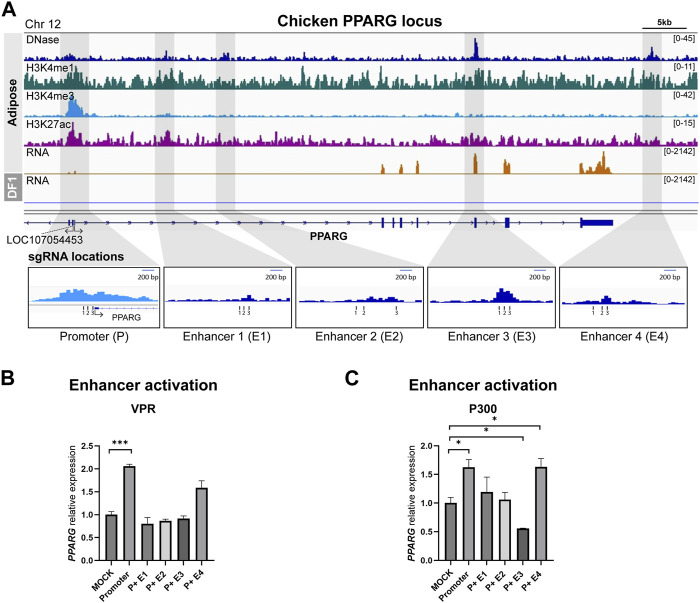
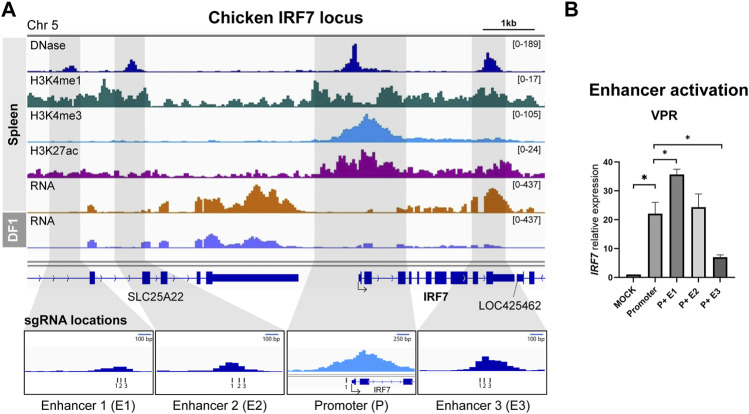
DNA regulatory elements intricately control when, where, and how genes are activated. Therefore, understanding the function of these elements could unveil the complexity of the genetic regulation network. Genome-wide significant variants are predominantly found in non-coding regions of DNA, so comprehending the predicted functional regulatory elements is crucial for understanding the biological context of these genomic markers, which can be incorporated into breeding programs. The emergence of CRISPR technology has provided a powerful tool for studying non-coding regulatory elements in genomes. In this study, we leveraged epigenetic data from the Functional Annotation of Animal Genomes project to identify promoter and putative enhancer regions associated with three genes (HBBA, IRF7, and PPARG) in the chicken genome. To identify the enhancer regions, we designed guide RNAs targeting the promoter and candidate enhancer regions and utilized CRISPR activation (CRISPRa) with dCas9-p300 and dCas9-VPR as transcriptional activators in chicken DF-1 cells. By comparing the expression levels of target genes between the promoter activation and the co-activation of the promoter and putative enhancers, we were able to identify functional enhancers that exhibited augmented upregulation. In conclusion, our findings demonstrate the remarkable efficiency of CRISPRa in precisely manipulating the expression of endogenous genes by targeting regulatory elements in the chicken genome, highlighting its potential for functional validation of non-coding regions.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
