Application of a new multi-locus variable number tandem repeat analysis (MLVA) scheme for the seasonal investigation of Cryptosporidium parvum cases in Wales and the northwest of England, spring 2022
Harriet Risby , Guy Robinson , Nastassya Chandra , Grace King , Roberto Vivancos , Robert Smith , Daniel Thomas , Andrew Fox , Noel McCarthy , Rachel M. Chalmers
{"title":"Application of a new multi-locus variable number tandem repeat analysis (MLVA) scheme for the seasonal investigation of Cryptosporidium parvum cases in Wales and the northwest of England, spring 2022","authors":"Harriet Risby , Guy Robinson , Nastassya Chandra , Grace King , Roberto Vivancos , Robert Smith , Daniel Thomas , Andrew Fox , Noel McCarthy , Rachel M. Chalmers","doi":"10.1016/j.crpvbd.2023.100151","DOIUrl":null,"url":null,"abstract":"<div><p>The protozoan <em>Cryptosporidium parvum</em> is an important cause of gastroenteritis in humans and livestock, and cryptosporidiosis outbreaks are common. However, a multi-locus genotyping scheme is not widely adopted. We describe the further development and application of a seven-locus multi-locus variable number of tandem repeats analysis (MLVA) scheme. From 28th March to 31st July 2022, confirmed <em>C. parvum</em> stools (<em>n</em> = 213) from cryptosporidiosis patients (cases) in Wales (<em>n</em> = 95) and the north west of England (<em>n</em> = 118) were tested by MLVA. Typability (defined as alleles identified at all seven loci in a sample) was 81.2% and discriminatory power estimated by Hunter Gaston Discriminatory Index was 0.99. A MLVA profile was constructed from the alleles, expressed in chromosomal order. Profiles were defined as simple (single allele at each locus) or mixed (more than one allele at any locus). A total of 161 MLVA profiles were identified; 13 were mixed, an additional 38 simple profiles contained null records, and 110 were complete simple profiles. A minimum spanning tree was constructed of simple MLVA profiles and those identical at all seven loci defined genetic clusters of cases (here, null records were considered as an allele); 77 cases formed 25 clusters, ranging from two to nine (mode = two) cases. The largest cluster, following epidemiological investigation, signalled a newly-identified outbreak. Two other cases with mixed profiles that contained the outbreak alleles were included in the outbreak investigation. In another epidemiologically-identified outbreak of six initial cases, MLVA detected two additional cases. In a third, small outbreak of three cases, identical MLVA profiles strengthened the microbiological evidence. Review of the performance characteristics of the individual loci and of the seven-locus scheme suggested that two loci might be candidates for review, but a larger dataset over a wider geographical area and longer timeframe will help inform decision-making about the scheme by user laboratories and stakeholders (such as public health agencies). This MLVA scheme is straightforward in use, fast and cheap compared to sequence-based methods, identifies mixed infections, provides an important tool for <em>C. parvum</em> surveillance, and can enhance outbreak investigations and public health action.</p></div>","PeriodicalId":94311,"journal":{"name":"Current research in parasitology & vector-borne diseases","volume":"4 ","pages":"Article 100151"},"PeriodicalIF":1.7000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2667114X23000390/pdfft?md5=4434a17c591dcde16d072e2f284155e9&pid=1-s2.0-S2667114X23000390-main.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current research in parasitology & vector-borne diseases","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667114X23000390","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/18 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"PARASITOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
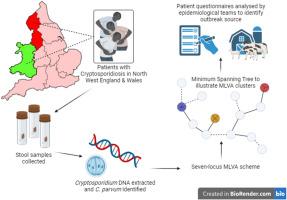
The protozoan Cryptosporidium parvum is an important cause of gastroenteritis in humans and livestock, and cryptosporidiosis outbreaks are common. However, a multi-locus genotyping scheme is not widely adopted. We describe the further development and application of a seven-locus multi-locus variable number of tandem repeats analysis (MLVA) scheme. From 28th March to 31st July 2022, confirmed C. parvum stools (n = 213) from cryptosporidiosis patients (cases) in Wales (n = 95) and the north west of England (n = 118) were tested by MLVA. Typability (defined as alleles identified at all seven loci in a sample) was 81.2% and discriminatory power estimated by Hunter Gaston Discriminatory Index was 0.99. A MLVA profile was constructed from the alleles, expressed in chromosomal order. Profiles were defined as simple (single allele at each locus) or mixed (more than one allele at any locus). A total of 161 MLVA profiles were identified; 13 were mixed, an additional 38 simple profiles contained null records, and 110 were complete simple profiles. A minimum spanning tree was constructed of simple MLVA profiles and those identical at all seven loci defined genetic clusters of cases (here, null records were considered as an allele); 77 cases formed 25 clusters, ranging from two to nine (mode = two) cases. The largest cluster, following epidemiological investigation, signalled a newly-identified outbreak. Two other cases with mixed profiles that contained the outbreak alleles were included in the outbreak investigation. In another epidemiologically-identified outbreak of six initial cases, MLVA detected two additional cases. In a third, small outbreak of three cases, identical MLVA profiles strengthened the microbiological evidence. Review of the performance characteristics of the individual loci and of the seven-locus scheme suggested that two loci might be candidates for review, but a larger dataset over a wider geographical area and longer timeframe will help inform decision-making about the scheme by user laboratories and stakeholders (such as public health agencies). This MLVA scheme is straightforward in use, fast and cheap compared to sequence-based methods, identifies mixed infections, provides an important tool for C. parvum surveillance, and can enhance outbreak investigations and public health action.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
