Anne R Shim, Kai Huang, Vadim Backman, Igal Szleifer
{"title":"Chromatin as self-returning walks: From population to single cell and back.","authors":"Anne R Shim, Kai Huang, Vadim Backman, Igal Szleifer","doi":"10.1016/j.bpr.2021.100042","DOIUrl":null,"url":null,"abstract":"<p><p>With a growing understanding of the chromatin structure, many efforts remain focused on bridging the gap between what is suggested by population-averaged data and what is visualized for single cells. A popular approach to traversing these scales is to fit a polymer model to Hi-C contact data. However, Hi-C is an average of millions to billions of cells, and each cell may not contain all population-averaged contacts. Therefore, we employ a novel approach of summing individual chromosome trajectories-determined by our Self-Returning Random Walk model-to create populations of cells. We allow single cells to consist of disparate structures and reproduce a variety of experimentally relevant contact maps. We show that the amount of shared topology between cells, and their mechanism of formation, changes the population-averaged structure. Therefore, we present a modeling technique that, with few constraints and little oversight, can be used to understand which single-cell chromatin structures underlie population-averaged behavior.</p>","PeriodicalId":72402,"journal":{"name":"Biophysical reports","volume":"2 1","pages":"100042"},"PeriodicalIF":2.7000,"publicationDate":"2022-03-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/24/c6/main.PMC9680733.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.bpr.2021.100042","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOPHYSICS","Score":null,"Total":0}
引用次数: 1
Abstract
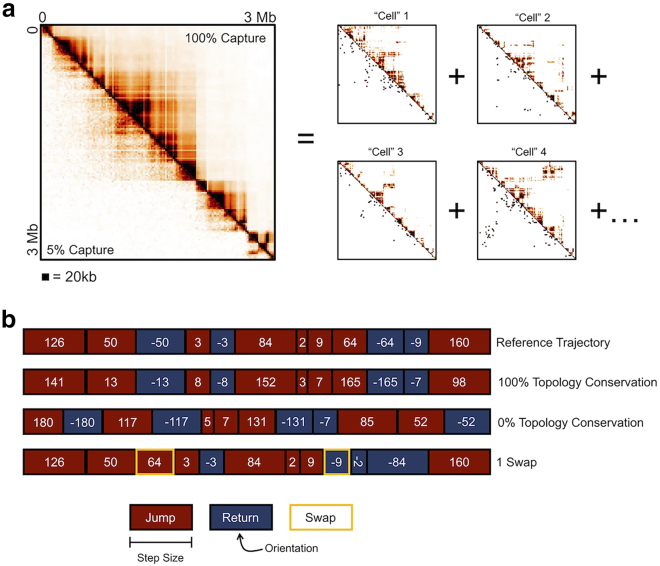
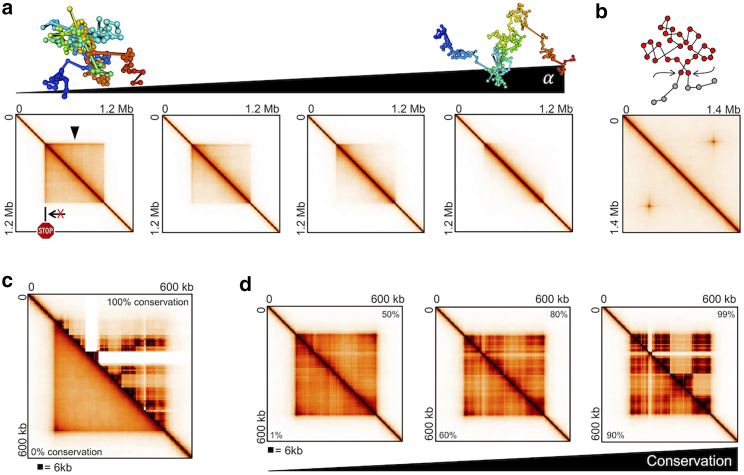
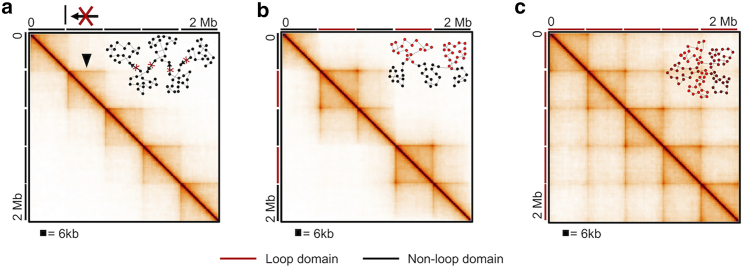
With a growing understanding of the chromatin structure, many efforts remain focused on bridging the gap between what is suggested by population-averaged data and what is visualized for single cells. A popular approach to traversing these scales is to fit a polymer model to Hi-C contact data. However, Hi-C is an average of millions to billions of cells, and each cell may not contain all population-averaged contacts. Therefore, we employ a novel approach of summing individual chromosome trajectories-determined by our Self-Returning Random Walk model-to create populations of cells. We allow single cells to consist of disparate structures and reproduce a variety of experimentally relevant contact maps. We show that the amount of shared topology between cells, and their mechanism of formation, changes the population-averaged structure. Therefore, we present a modeling technique that, with few constraints and little oversight, can be used to understand which single-cell chromatin structures underlie population-averaged behavior.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
