Gabriel E Hoffman, Donghoon Lee, Jaroslav Bendl, N M Prashant, Aram Hong, Clara Casey, Marcela Alvia, Zhiping Shao, Stathis Argyriou, Karen Therrien, Sanan Venkatesh, Georgios Voloudakis, Vahram Haroutunian, John F Fullard, Panos Roussos
{"title":"Efficient differential expression analysis of large-scale single cell transcriptomics data using dreamlet.","authors":"Gabriel E Hoffman, Donghoon Lee, Jaroslav Bendl, N M Prashant, Aram Hong, Clara Casey, Marcela Alvia, Zhiping Shao, Stathis Argyriou, Karen Therrien, Sanan Venkatesh, Georgios Voloudakis, Vahram Haroutunian, John F Fullard, Panos Roussos","doi":"10.1101/2023.03.17.533005","DOIUrl":null,"url":null,"abstract":"<p><p>Advances in single-cell and -nucleus transcriptomics have enabled generation of increasingly large-scale datasets from hundreds of subjects and millions of cells. These studies promise to give unprecedented insight into the cell type specific biology of human disease. Yet performing differential expression analyses across subjects remains difficult due to challenges in statistical modeling of these complex studies and scaling analyses to large datasets. Our open-source R package dreamlet (DiseaseNeurogenomics.github.io/dreamlet) uses a pseudobulk approach based on precision-weighted linear mixed models to identify genes differentially expressed with traits across subjects for each cell cluster. Designed for data from large cohorts, dreamlet is substantially faster and uses less memory than existing workflows, while supporting complex statistical models and controlling the false positive rate. We demonstrate computational and statistical performance on published datasets, and a novel dataset of 1.4M single nuclei from postmortem brains of 150 Alzheimer's disease cases and 149 controls.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/56/f1/nihpp-2023.03.17.533005v1.PMC10055252.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.03.17.533005","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
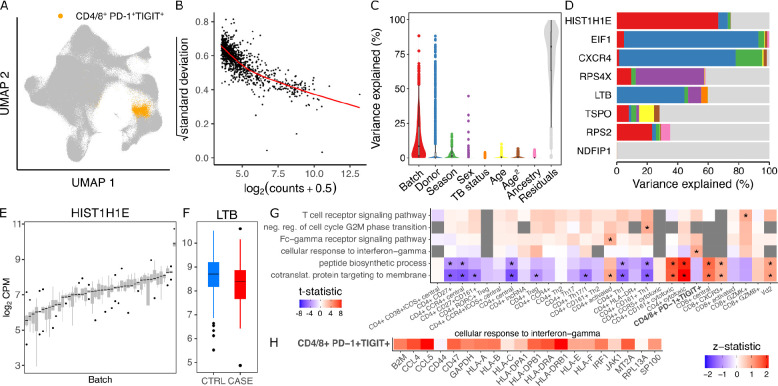
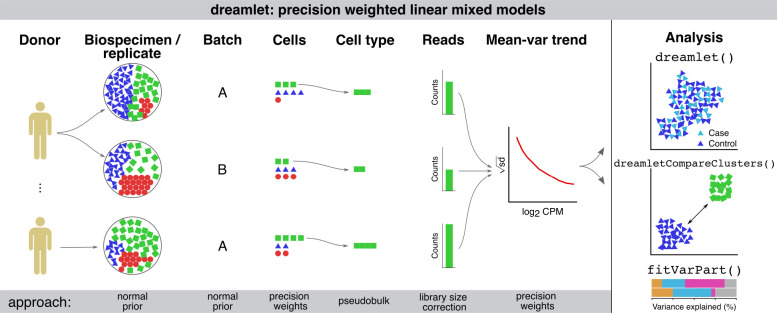
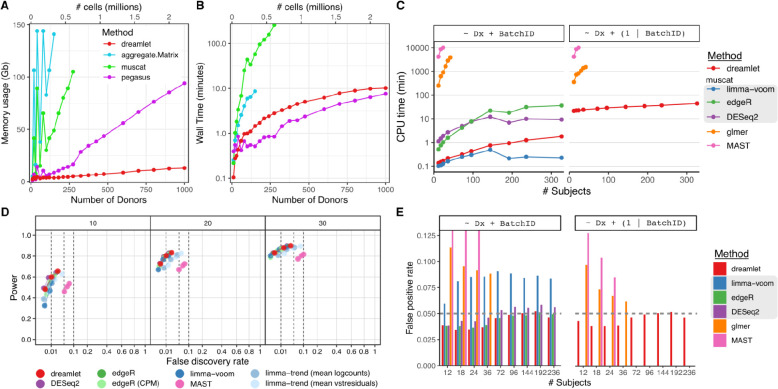
Advances in single-cell and -nucleus transcriptomics have enabled generation of increasingly large-scale datasets from hundreds of subjects and millions of cells. These studies promise to give unprecedented insight into the cell type specific biology of human disease. Yet performing differential expression analyses across subjects remains difficult due to challenges in statistical modeling of these complex studies and scaling analyses to large datasets. Our open-source R package dreamlet (DiseaseNeurogenomics.github.io/dreamlet) uses a pseudobulk approach based on precision-weighted linear mixed models to identify genes differentially expressed with traits across subjects for each cell cluster. Designed for data from large cohorts, dreamlet is substantially faster and uses less memory than existing workflows, while supporting complex statistical models and controlling the false positive rate. We demonstrate computational and statistical performance on published datasets, and a novel dataset of 1.4M single nuclei from postmortem brains of 150 Alzheimer's disease cases and 149 controls.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
