Nicole V Dowling, Todd A Naumann, Neil P J Price, David R Rose
{"title":"Crystal structure of a polyglycine hydrolase determined using a RoseTTAFold model.","authors":"Nicole V Dowling, Todd A Naumann, Neil P J Price, David R Rose","doi":"10.1107/S2059798323000311","DOIUrl":null,"url":null,"abstract":"<p><p>Polyglycine hydrolases (PGHs) are secreted fungal proteases that cleave the polyglycine linker of Zea mays ChitA, a defensive chitinase, thus overcoming one mechanism of plant resistance to infection. Despite their importance in agriculture, there has been no previous structural characterization of this family of proteases. The objective of this research was to investigate the proteolytic mechanism and other characteristics by structural and biochemical means. Here, the first atomic structure of a polyglycine hydrolase was identified. It was solved by X-ray crystallography using a RoseTTAFold model, taking advantage of recent technical advances in structure prediction. PGHs are composed of two domains: the N- and C-domains. The N-domain is a novel tertiary fold with an as-yet unknown function that is found across all kingdoms of life. The C-domain shares structural similarities with class C β-lactamases, including a common catalytic nucleophilic serine. In addition to insights into the PGH family and its relationship to β-lactamases, the results demonstrate the power of complementing experimental structure determination with new computational techniques.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":"79 Pt 2","pages":"168-176"},"PeriodicalIF":3.8000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9912923/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798323000311","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/2/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
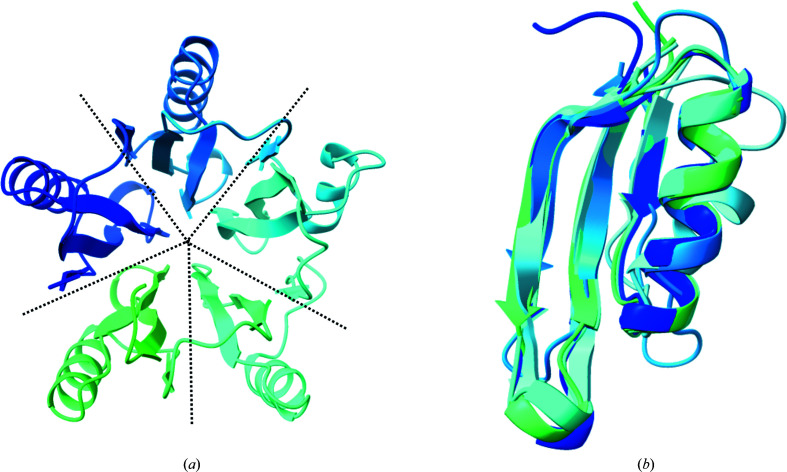
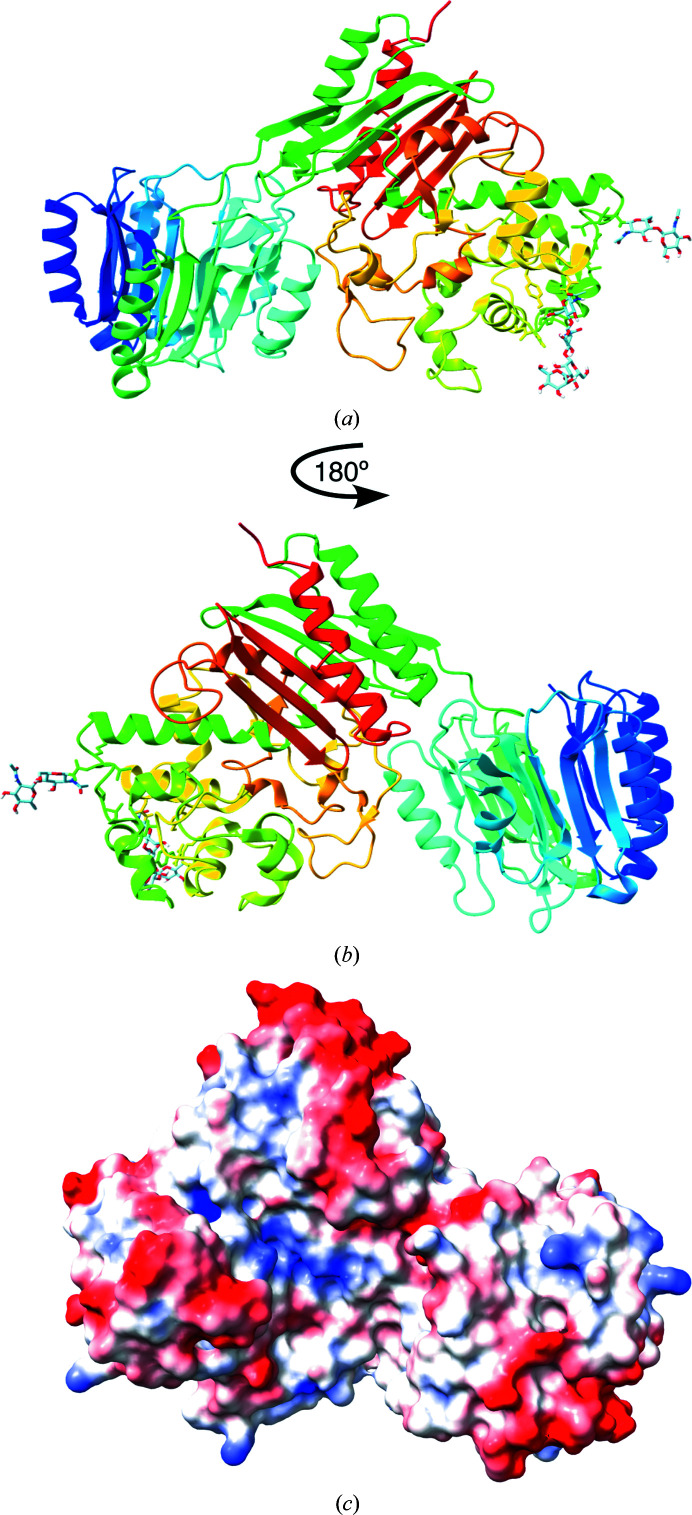
Polyglycine hydrolases (PGHs) are secreted fungal proteases that cleave the polyglycine linker of Zea mays ChitA, a defensive chitinase, thus overcoming one mechanism of plant resistance to infection. Despite their importance in agriculture, there has been no previous structural characterization of this family of proteases. The objective of this research was to investigate the proteolytic mechanism and other characteristics by structural and biochemical means. Here, the first atomic structure of a polyglycine hydrolase was identified. It was solved by X-ray crystallography using a RoseTTAFold model, taking advantage of recent technical advances in structure prediction. PGHs are composed of two domains: the N- and C-domains. The N-domain is a novel tertiary fold with an as-yet unknown function that is found across all kingdoms of life. The C-domain shares structural similarities with class C β-lactamases, including a common catalytic nucleophilic serine. In addition to insights into the PGH family and its relationship to β-lactamases, the results demonstrate the power of complementing experimental structure determination with new computational techniques.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
