{"title":"Effects of Antibiotics on the Uterine Microbial Community of Mice.","authors":"Sang-Gyu Kim, Dae-Wi Kim, Hoon Jang","doi":"10.12717/DR.2022.26.4.145","DOIUrl":null,"url":null,"abstract":"<p><p>The gut microbiota is involved in the maintenance of physiological homeostasis and is now recognized as a regulator of many diseases. Although germ-free mouse models are the standard for microbiome studies, mice with antibiotic-induced sterile intestines are often chosen as a fast and inexpensive alternative. Pathophysiological changes in the gut microbiome have been demonstrated, but there are no reports so far on how such alterations affect the bacterial composition of the uterus. Here we examined changes in uterine microbiota as a result of gut microbiome disruption in an antibiotics-based sterile-uterus mouse model. Sterility was induced in 6-week-old female mice by administration of a combination of antibiotics, and amplicons of a bacteria marker gene (16S rRNA) were sequenced to decipher bacterial community structures in the uterus. At the phylum-level, Proteobacteria, Firmicutes, and Actinobacteria were found to be dominant, while <i>Ralstonia</i>, <i>Escherichia</i>, and <i>Prauserella</i> were the major genera. Quantitative comparisons of the microbial contents of an antibiotic-fed and a control group revealed that the treatment resulted in the reduction of bacterial population density. Although there was no significant difference in bacterial community structures between the two animal groups, β-diversity analysis showed a converged profile of uterus microbiotain the germ-free model. These findings suggest that the induction of sterility does not result in changes in the levels of specific taxa but in a reduction of individual variations in the mouse uterus microbiota, accompanied by a decrease in overall bacterial population density.</p>","PeriodicalId":72791,"journal":{"name":"Development & reproduction","volume":"26 4","pages":"145-153"},"PeriodicalIF":0.0000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/99/10/dr-26-4-145.PMC9925184.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Development & reproduction","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.12717/DR.2022.26.4.145","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
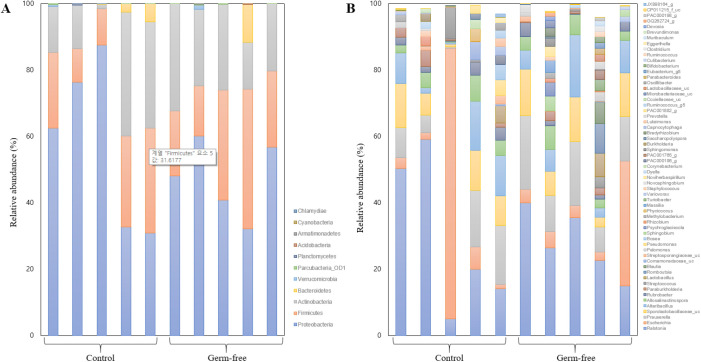
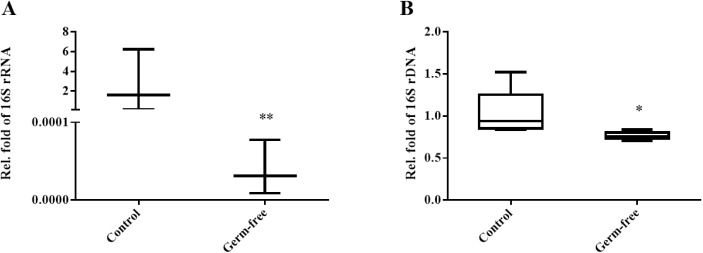
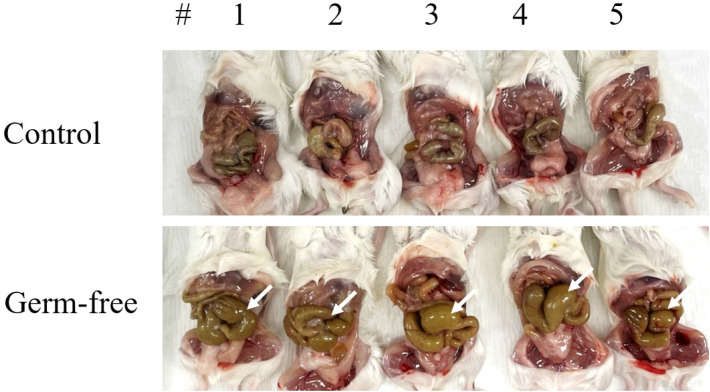
The gut microbiota is involved in the maintenance of physiological homeostasis and is now recognized as a regulator of many diseases. Although germ-free mouse models are the standard for microbiome studies, mice with antibiotic-induced sterile intestines are often chosen as a fast and inexpensive alternative. Pathophysiological changes in the gut microbiome have been demonstrated, but there are no reports so far on how such alterations affect the bacterial composition of the uterus. Here we examined changes in uterine microbiota as a result of gut microbiome disruption in an antibiotics-based sterile-uterus mouse model. Sterility was induced in 6-week-old female mice by administration of a combination of antibiotics, and amplicons of a bacteria marker gene (16S rRNA) were sequenced to decipher bacterial community structures in the uterus. At the phylum-level, Proteobacteria, Firmicutes, and Actinobacteria were found to be dominant, while Ralstonia, Escherichia, and Prauserella were the major genera. Quantitative comparisons of the microbial contents of an antibiotic-fed and a control group revealed that the treatment resulted in the reduction of bacterial population density. Although there was no significant difference in bacterial community structures between the two animal groups, β-diversity analysis showed a converged profile of uterus microbiotain the germ-free model. These findings suggest that the induction of sterility does not result in changes in the levels of specific taxa but in a reduction of individual variations in the mouse uterus microbiota, accompanied by a decrease in overall bacterial population density.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
