Shruti Menon, Daniel Gracilla, Marcus R Breese, Yone Phar Lin, Filemon Dela Cruz, Tamar Feinberg, Elisa de Stanchina, Ana-Florina Galic, Hannah Allegakoen, Shruthi Perati, Nicholas Wen, Ann Heslin, Max A Horlbeck, Jonathan Weissman, E Alejandro Sweet-Cordero, Trever G Bivona, Asmin Tulpule
{"title":"FET fusion oncoproteins disrupt physiologic DNA repair and create a targetable opportunity for ATR inhibitor therapy.","authors":"Shruti Menon, Daniel Gracilla, Marcus R Breese, Yone Phar Lin, Filemon Dela Cruz, Tamar Feinberg, Elisa de Stanchina, Ana-Florina Galic, Hannah Allegakoen, Shruthi Perati, Nicholas Wen, Ann Heslin, Max A Horlbeck, Jonathan Weissman, E Alejandro Sweet-Cordero, Trever G Bivona, Asmin Tulpule","doi":"10.1101/2023.04.30.538578","DOIUrl":null,"url":null,"abstract":"<p><p>In cancers with genetic loss of specific DNA damage response (DDR) genes (i.e., BRCA1/2 tumor suppressor mutations), synthetic lethal targeting of compensatory DDR pathways has translated into clinical benefit for patients. Whether and how growth-promoting oncogenes might also create tumor-specific vulnerabilities within DDR networks is not well understood. Here we focus on Ewing sarcoma, a FET fusion oncoprotein (EWSR1-FLI1) driven pediatric bone tumor, as a model for the class of FET rearranged cancers. Native FET family members are among the earliest factors recruited to DNA double-strand breaks (DSBs), though the function of both native FET proteins and FET fusion oncoproteins in DNA repair remains to be defined. We discover that EWSR1-FLI1 and other FET fusion oncoproteins are recruited to DNA DSBs and impair the activation and downstream signaling of the DNA damage sensor ATM. In multiple FET rearranged cancers, we establish the compensatory ATR signaling axis as a collateral dependency and therapeutic target using patient-derived xenograft models. In summary, we describe how oncogenes can disrupt physiologic DNA repair and provide the preclinical rationale for specifically testing ATR inhibitors in FET rearranged cancers as part of ongoing early phase clinical trials.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-05-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/db/nihpp-2023.04.30.538578v2.PMC10187251.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.04.30.538578","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
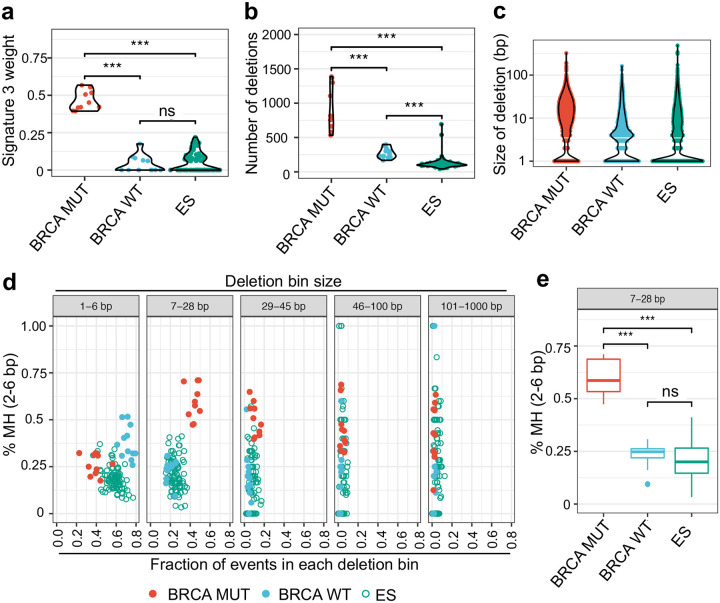
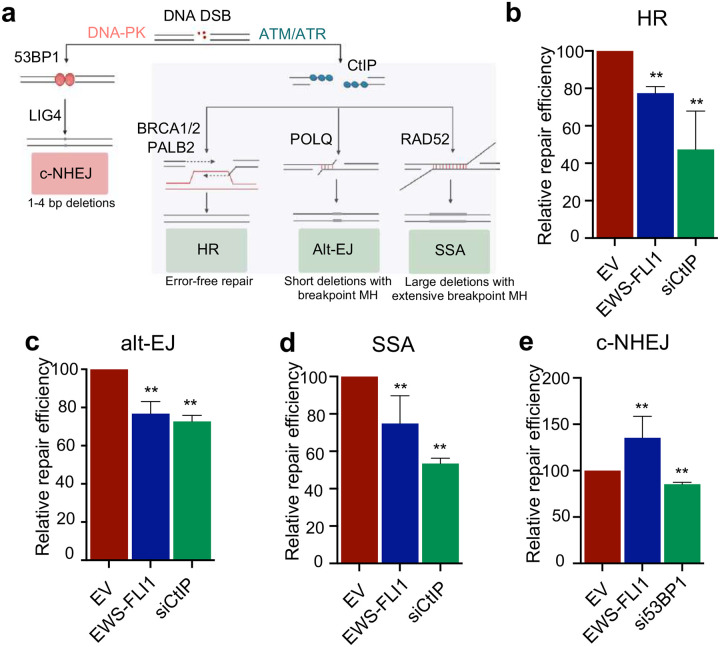
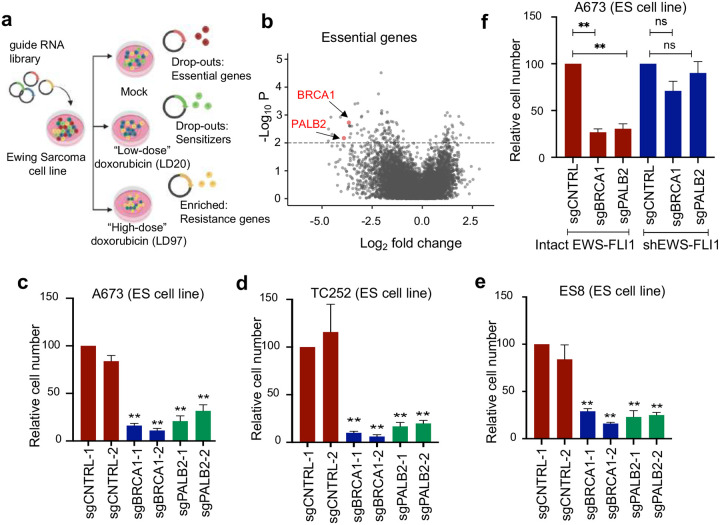
In cancers with genetic loss of specific DNA damage response (DDR) genes (i.e., BRCA1/2 tumor suppressor mutations), synthetic lethal targeting of compensatory DDR pathways has translated into clinical benefit for patients. Whether and how growth-promoting oncogenes might also create tumor-specific vulnerabilities within DDR networks is not well understood. Here we focus on Ewing sarcoma, a FET fusion oncoprotein (EWSR1-FLI1) driven pediatric bone tumor, as a model for the class of FET rearranged cancers. Native FET family members are among the earliest factors recruited to DNA double-strand breaks (DSBs), though the function of both native FET proteins and FET fusion oncoproteins in DNA repair remains to be defined. We discover that EWSR1-FLI1 and other FET fusion oncoproteins are recruited to DNA DSBs and impair the activation and downstream signaling of the DNA damage sensor ATM. In multiple FET rearranged cancers, we establish the compensatory ATR signaling axis as a collateral dependency and therapeutic target using patient-derived xenograft models. In summary, we describe how oncogenes can disrupt physiologic DNA repair and provide the preclinical rationale for specifically testing ATR inhibitors in FET rearranged cancers as part of ongoing early phase clinical trials.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
