Online Phylogenetics with matOptimize Produces Equivalent Trees and is Dramatically More Efficient for Large SARS-CoV-2 Phylogenies than de novo and Maximum-Likelihood Implementations.
Alexander M Kramer, Bryan Thornlow, Cheng Ye, Nicola De Maio, Jakob McBroome, Angie S Hinrichs, Robert Lanfear, Yatish Turakhia, Russell Corbett-Detig
{"title":"Online Phylogenetics with matOptimize Produces Equivalent Trees and is Dramatically More Efficient for Large SARS-CoV-2 Phylogenies than de novo and Maximum-Likelihood Implementations.","authors":"Alexander M Kramer, Bryan Thornlow, Cheng Ye, Nicola De Maio, Jakob McBroome, Angie S Hinrichs, Robert Lanfear, Yatish Turakhia, Russell Corbett-Detig","doi":"10.1093/sysbio/syad031","DOIUrl":null,"url":null,"abstract":"<p><p>Phylogenetics has been foundational to SARS-CoV-2 research and public health policy, assisting in genomic surveillance, contact tracing, and assessing emergence and spread of new variants. However, phylogenetic analyses of SARS-CoV-2 have often relied on tools designed for de novo phylogenetic inference, in which all data are collected before any analysis is performed and the phylogeny is inferred once from scratch. SARS-CoV-2 data sets do not fit this mold. There are currently over 14 million sequenced SARS-CoV-2 genomes in online databases, with tens of thousands of new genomes added every day. Continuous data collection, combined with the public health relevance of SARS-CoV-2, invites an \"online\" approach to phylogenetics, in which new samples are added to existing phylogenetic trees every day. The extremely dense sampling of SARS-CoV-2 genomes also invites a comparison between likelihood and parsimony approaches to phylogenetic inference. Maximum likelihood (ML) and pseudo-ML methods may be more accurate when there are multiple changes at a single site on a single branch, but this accuracy comes at a large computational cost, and the dense sampling of SARS-CoV-2 genomes means that these instances will be extremely rare because each internal branch is expected to be extremely short. Therefore, it may be that approaches based on maximum parsimony (MP) are sufficiently accurate for reconstructing phylogenies of SARS-CoV-2, and their simplicity means that they can be applied to much larger data sets. Here, we evaluate the performance of de novo and online phylogenetic approaches, as well as ML, pseudo-ML, and MP frameworks for inferring large and dense SARS-CoV-2 phylogenies. Overall, we find that online phylogenetics produces similar phylogenetic trees to de novo analyses for SARS-CoV-2, and that MP optimization with UShER and matOptimize produces equivalent SARS-CoV-2 phylogenies to some of the most popular ML and pseudo-ML inference tools. MP optimization with UShER and matOptimize is thousands of times faster than presently available implementations of ML and online phylogenetics is faster than de novo inference. Our results therefore suggest that parsimony-based methods like UShER and matOptimize represent an accurate and more practical alternative to established ML implementations for large SARS-CoV-2 phylogenies and could be successfully applied to other similar data sets with particularly dense sampling and short branch lengths.</p>","PeriodicalId":22120,"journal":{"name":"Systematic Biology","volume":" ","pages":"1039-1051"},"PeriodicalIF":5.7000,"publicationDate":"2023-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10627557/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Systematic Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/sysbio/syad031","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
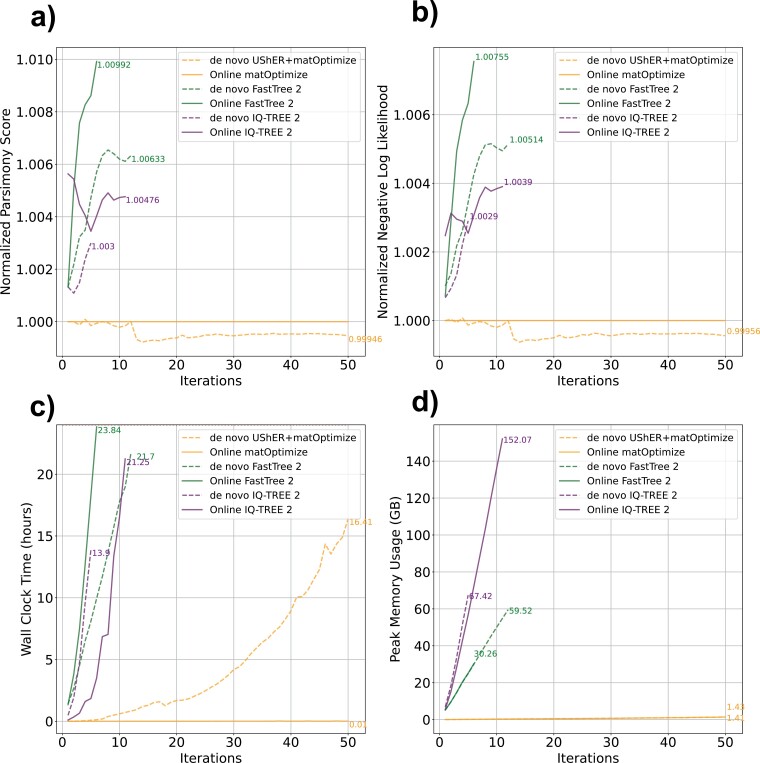
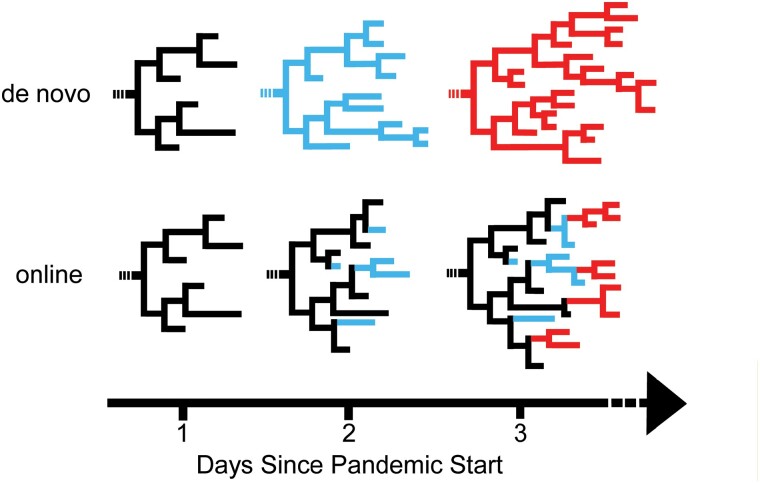
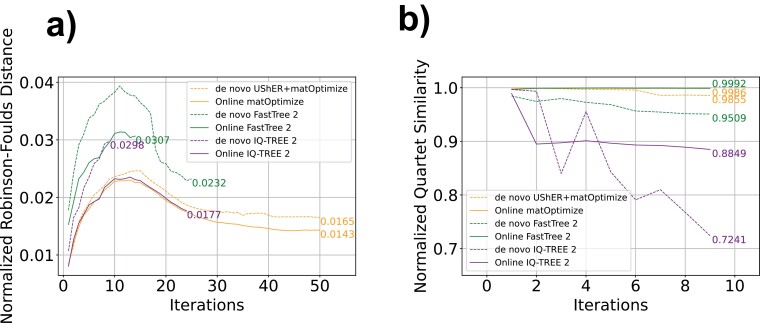
Phylogenetics has been foundational to SARS-CoV-2 research and public health policy, assisting in genomic surveillance, contact tracing, and assessing emergence and spread of new variants. However, phylogenetic analyses of SARS-CoV-2 have often relied on tools designed for de novo phylogenetic inference, in which all data are collected before any analysis is performed and the phylogeny is inferred once from scratch. SARS-CoV-2 data sets do not fit this mold. There are currently over 14 million sequenced SARS-CoV-2 genomes in online databases, with tens of thousands of new genomes added every day. Continuous data collection, combined with the public health relevance of SARS-CoV-2, invites an "online" approach to phylogenetics, in which new samples are added to existing phylogenetic trees every day. The extremely dense sampling of SARS-CoV-2 genomes also invites a comparison between likelihood and parsimony approaches to phylogenetic inference. Maximum likelihood (ML) and pseudo-ML methods may be more accurate when there are multiple changes at a single site on a single branch, but this accuracy comes at a large computational cost, and the dense sampling of SARS-CoV-2 genomes means that these instances will be extremely rare because each internal branch is expected to be extremely short. Therefore, it may be that approaches based on maximum parsimony (MP) are sufficiently accurate for reconstructing phylogenies of SARS-CoV-2, and their simplicity means that they can be applied to much larger data sets. Here, we evaluate the performance of de novo and online phylogenetic approaches, as well as ML, pseudo-ML, and MP frameworks for inferring large and dense SARS-CoV-2 phylogenies. Overall, we find that online phylogenetics produces similar phylogenetic trees to de novo analyses for SARS-CoV-2, and that MP optimization with UShER and matOptimize produces equivalent SARS-CoV-2 phylogenies to some of the most popular ML and pseudo-ML inference tools. MP optimization with UShER and matOptimize is thousands of times faster than presently available implementations of ML and online phylogenetics is faster than de novo inference. Our results therefore suggest that parsimony-based methods like UShER and matOptimize represent an accurate and more practical alternative to established ML implementations for large SARS-CoV-2 phylogenies and could be successfully applied to other similar data sets with particularly dense sampling and short branch lengths.
期刊介绍:
Systematic Biology is the bimonthly journal of the Society of Systematic Biologists. Papers for the journal are original contributions to the theory, principles, and methods of systematics as well as phylogeny, evolution, morphology, biogeography, paleontology, genetics, and the classification of all living things. A Points of View section offers a forum for discussion, while book reviews and announcements of general interest are also featured.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
