Mohammad Salma, Elina Alaterre, Jérôme Moreaux, Eric Soler
{"title":"Var∣Decrypt: a novel and user-friendly tool to explore and prioritize variants in whole-exome sequencing data.","authors":"Mohammad Salma, Elina Alaterre, Jérôme Moreaux, Eric Soler","doi":"10.1186/s13072-023-00497-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>High-throughput sequencing (HTS) offers unprecedented opportunities for the discovery of causative gene variants in multiple human disorders including cancers, and has revolutionized clinical diagnostics. However, despite more than a decade of use of HTS-based assays, extracting relevant functional information from whole-exome sequencing (WES) data remains challenging, especially for non-specialists lacking in-depth bioinformatic skills.</p><p><strong>Results: </strong>To address this limitation, we developed Var∣Decrypt, a web-based tool designed to greatly facilitate WES data browsing and analysis. Var∣Decrypt offers a wide range of gene and variant filtering possibilities, clustering and enrichment tools, providing an efficient way to derive patient-specific functional information and to prioritize gene variants for functional analyses. We applied Var∣Decrypt on WES datasets of 10 acute erythroid leukemia patients, a rare and aggressive form of leukemia, and recovered known disease oncogenes in addition to novel putative drivers. We additionally validated the performance of Var∣Decrypt using an independent dataset of ~ 90 multiple myeloma WES, recapitulating the identified deregulated genes and pathways, showing the general applicability and versatility of Var∣Decrypt for WES analysis.</p><p><strong>Conclusion: </strong>Despite years of use of WES in human health for diagnosis and discovery of disease drivers, WES data analysis still remains a complex task requiring advanced bioinformatic skills. In that context, there is a need for user-friendly all-in-one dedicated tools for data analysis, to allow biologists and clinicians to extract relevant biological information from patient datasets. Here, we provide Var∣Decrypt (trial version accessible here: https://vardecrypt.com/app/vardecrypt ), a simple and intuitive Rshiny application created to fill this gap. Source code and detailed user tutorial are available at https://gitlab.com/mohammadsalma/vardecrypt .</p>","PeriodicalId":49253,"journal":{"name":"Epigenetics & Chromatin","volume":"16 1","pages":"23"},"PeriodicalIF":3.5000,"publicationDate":"2023-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10265870/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epigenetics & Chromatin","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13072-023-00497-4","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
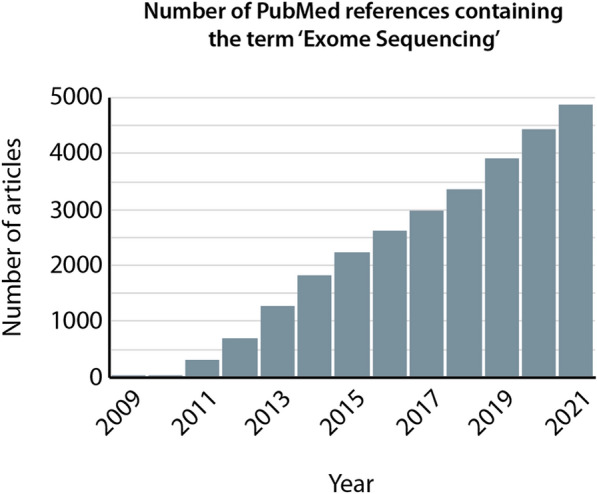
Background: High-throughput sequencing (HTS) offers unprecedented opportunities for the discovery of causative gene variants in multiple human disorders including cancers, and has revolutionized clinical diagnostics. However, despite more than a decade of use of HTS-based assays, extracting relevant functional information from whole-exome sequencing (WES) data remains challenging, especially for non-specialists lacking in-depth bioinformatic skills.
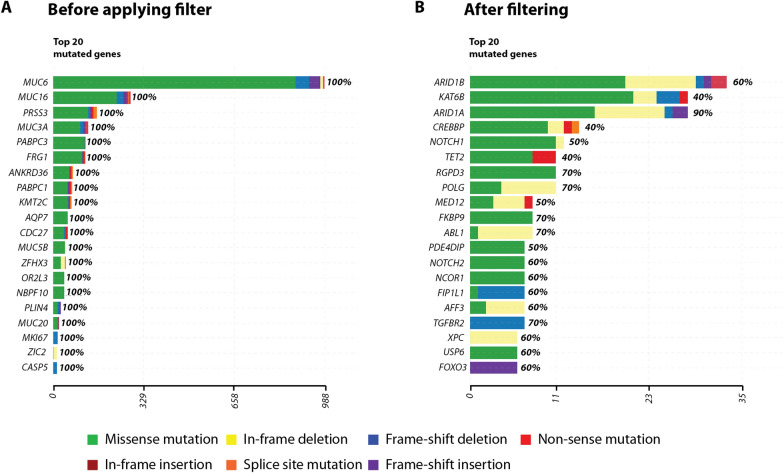
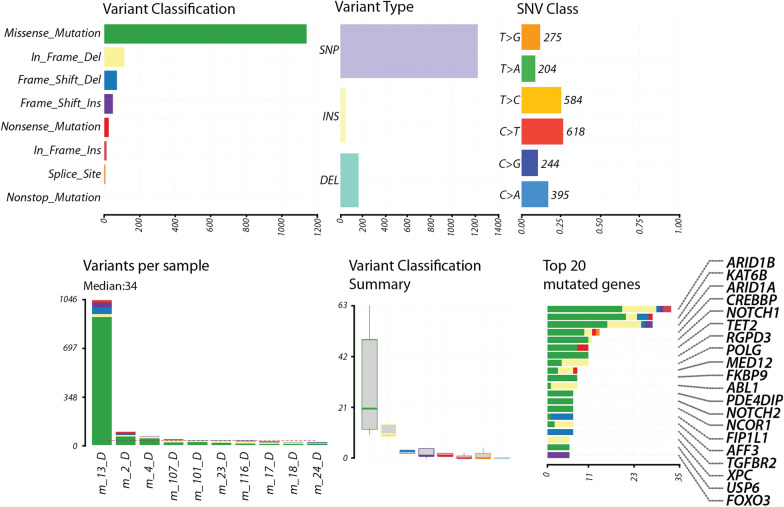
Results: To address this limitation, we developed Var∣Decrypt, a web-based tool designed to greatly facilitate WES data browsing and analysis. Var∣Decrypt offers a wide range of gene and variant filtering possibilities, clustering and enrichment tools, providing an efficient way to derive patient-specific functional information and to prioritize gene variants for functional analyses. We applied Var∣Decrypt on WES datasets of 10 acute erythroid leukemia patients, a rare and aggressive form of leukemia, and recovered known disease oncogenes in addition to novel putative drivers. We additionally validated the performance of Var∣Decrypt using an independent dataset of ~ 90 multiple myeloma WES, recapitulating the identified deregulated genes and pathways, showing the general applicability and versatility of Var∣Decrypt for WES analysis.
Conclusion: Despite years of use of WES in human health for diagnosis and discovery of disease drivers, WES data analysis still remains a complex task requiring advanced bioinformatic skills. In that context, there is a need for user-friendly all-in-one dedicated tools for data analysis, to allow biologists and clinicians to extract relevant biological information from patient datasets. Here, we provide Var∣Decrypt (trial version accessible here: https://vardecrypt.com/app/vardecrypt ), a simple and intuitive Rshiny application created to fill this gap. Source code and detailed user tutorial are available at https://gitlab.com/mohammadsalma/vardecrypt .
期刊介绍:
Epigenetics & Chromatin is a peer-reviewed, open access, online journal that publishes research, and reviews, providing novel insights into epigenetic inheritance and chromatin-based interactions. The journal aims to understand how gene and chromosomal elements are regulated and their activities maintained during processes such as cell division, differentiation and environmental alteration.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
