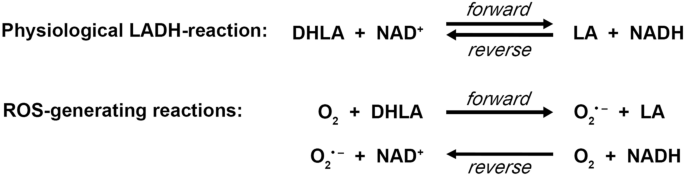
{"title":"Lipoamide dehydrogenase (LADH) deficiency: medical perspectives of the structural and functional characterization of LADH and its pathogenic variants.","authors":"Eszter Szabó, Attila Ambrus","doi":"10.1007/s42977-023-00155-6","DOIUrl":null,"url":null,"abstract":"<p><p>(Dihydro)lipoamide dehydrogenase (LADH) deficiency is an autosomal recessive genetic metabolic disorder. It generally presents with an onset in the neonatal age and premature death. The clinical picture usually involves metabolic decompensation and lactic acidosis that lead to neurological, cardiological, and/or hepatological outcomes. Severity of the disease is due to the fact that LADH is a common E3 subunit to the pyruvate, alpha-ketoglutarate, alpha-ketoadipate, and branched-chain alpha-keto acid dehydrogenase complexes and is also part of the glycine cleavage system; hence, a loss in LADH activity adversely affects several central metabolic pathways simultaneously. The severe clinical manifestations, however, often do not parallel the LADH activity loss, which implies the existence of auxiliary pathological pathways; stimulated reactive oxygen species (ROS) production as well as dissociation from the relevant multienzyme complexes proved to be auxiliary exacerbating pathomechanisms for selected disease-causing LADH mutations. This review provides an overview on the therapeutic challenges of inherited metabolic diseases, structural and functional characteristics of the mitochondrial alpha-keto acid dehydrogenase complexes, molecular pathogenesis and structural basis of LADH deficiency, and relevant potential future medical perspectives.</p>","PeriodicalId":8853,"journal":{"name":"Biologia futura","volume":"74 1-2","pages":"109-118"},"PeriodicalIF":1.5000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biologia futura","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s42977-023-00155-6","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
(Dihydro)lipoamide dehydrogenase (LADH) deficiency is an autosomal recessive genetic metabolic disorder. It generally presents with an onset in the neonatal age and premature death. The clinical picture usually involves metabolic decompensation and lactic acidosis that lead to neurological, cardiological, and/or hepatological outcomes. Severity of the disease is due to the fact that LADH is a common E3 subunit to the pyruvate, alpha-ketoglutarate, alpha-ketoadipate, and branched-chain alpha-keto acid dehydrogenase complexes and is also part of the glycine cleavage system; hence, a loss in LADH activity adversely affects several central metabolic pathways simultaneously. The severe clinical manifestations, however, often do not parallel the LADH activity loss, which implies the existence of auxiliary pathological pathways; stimulated reactive oxygen species (ROS) production as well as dissociation from the relevant multienzyme complexes proved to be auxiliary exacerbating pathomechanisms for selected disease-causing LADH mutations. This review provides an overview on the therapeutic challenges of inherited metabolic diseases, structural and functional characteristics of the mitochondrial alpha-keto acid dehydrogenase complexes, molecular pathogenesis and structural basis of LADH deficiency, and relevant potential future medical perspectives.
Biologia futuraAgricultural and Biological Sciences-Agricultural and Biological Sciences (all)
CiteScore
3.50
自引率
0.00%
发文量
27
期刊介绍:
How can the scientific knowledge we possess now influence that future? That is, the FUTURE of Earth and life − of humankind. Can we make choices in the present to change our future? How can 21st century biological research ask proper scientific questions and find solid answers? Addressing these questions is the main goal of Biologia Futura (formerly Acta Biologica Hungarica).
In keeping with the name, the new mission is to focus on areas of biology where major advances are to be expected, areas of biology with strong inter-disciplinary connection and to provide new avenues for future research in biology. Biologia Futura aims to publish articles from all fields of biology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
