Sara Fañanas-Baquero, Matías Morín, Sergio Fernández, Isabel Ojeda-Perez, Mercedes Dessy-Rodriguez, Miruna Giurgiu, Juan A Bueren, Miguel Angel Moreno-Pelayo, Jose Carlos Segovia, Oscar Quintana-Bustamante
{"title":"Specific correction of pyruvate kinase deficiency-causing point mutations by CRISPR/Cas9 and single-stranded oligodeoxynucleotides.","authors":"Sara Fañanas-Baquero, Matías Morín, Sergio Fernández, Isabel Ojeda-Perez, Mercedes Dessy-Rodriguez, Miruna Giurgiu, Juan A Bueren, Miguel Angel Moreno-Pelayo, Jose Carlos Segovia, Oscar Quintana-Bustamante","doi":"10.3389/fgeed.2023.1104666","DOIUrl":null,"url":null,"abstract":"<p><p>Pyruvate kinase deficiency (PKD) is an autosomal recessive disorder caused by mutations in the <i>PKLR</i> gene. PKD-erythroid cells suffer from an energy imbalance caused by a reduction of erythroid pyruvate kinase (RPK) enzyme activity. PKD is associated with reticulocytosis, splenomegaly and iron overload, and may be life-threatening in severely affected patients. More than 300 disease-causing mutations have been identified as causing PKD. Most mutations are missense mutations, commonly present as compound heterozygous. Therefore, specific correction of these point mutations might be a promising therapy for the treatment of PKD patients. We have explored the potential of precise gene editing for the correction of different PKD-causing mutations, using a combination of single-stranded oligodeoxynucleotides (ssODN) with the CRISPR/Cas9 system. We have designed guide RNAs (gRNAs) and single-strand donor templates to target four different PKD-causing mutations in immortalized patient-derived lymphoblastic cell lines, and we have detected the precise correction in three of these mutations. The frequency of the precise gene editing is variable, while the presence of additional insertions/deletions (InDels) has also been detected. Significantly, we have identified high mutation-specificity for two of the PKD-causing mutations. Our results demonstrate the feasibility of a highly personalized gene-editing therapy to treat point mutations in cells derived from PKD patients.</p>","PeriodicalId":73086,"journal":{"name":"Frontiers in genome editing","volume":"5 ","pages":"1104666"},"PeriodicalIF":4.4000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10175809/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in genome editing","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fgeed.2023.1104666","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
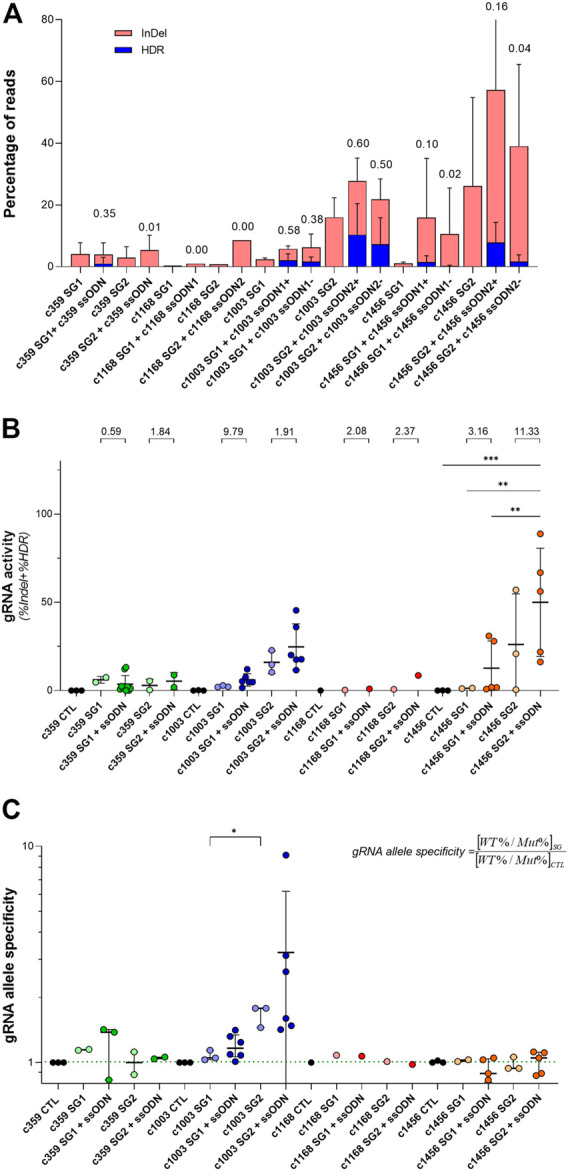
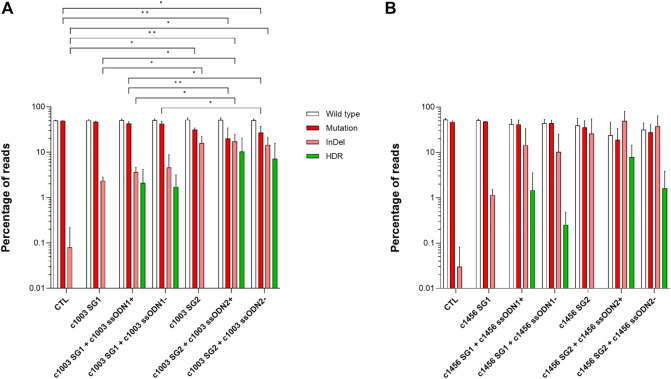
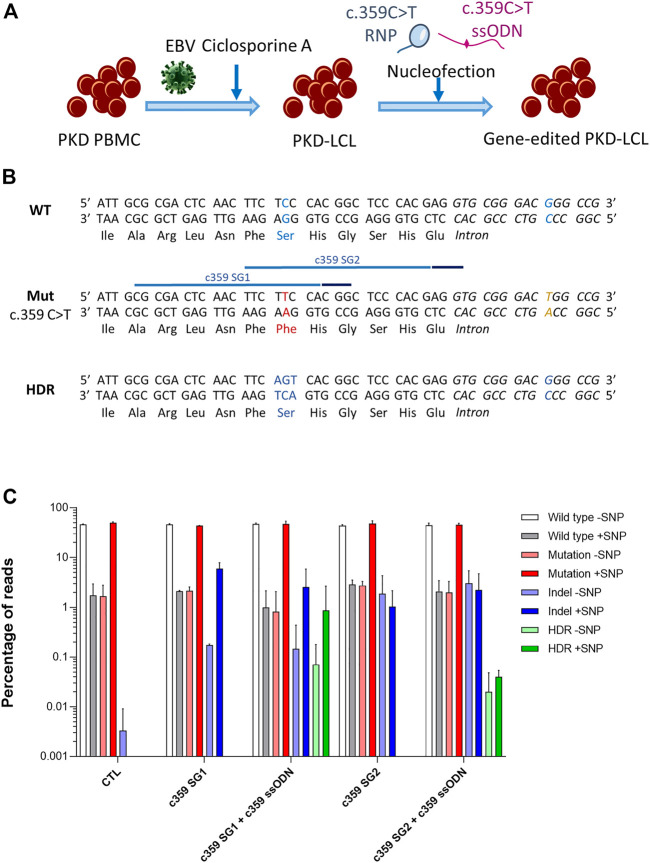
Pyruvate kinase deficiency (PKD) is an autosomal recessive disorder caused by mutations in the PKLR gene. PKD-erythroid cells suffer from an energy imbalance caused by a reduction of erythroid pyruvate kinase (RPK) enzyme activity. PKD is associated with reticulocytosis, splenomegaly and iron overload, and may be life-threatening in severely affected patients. More than 300 disease-causing mutations have been identified as causing PKD. Most mutations are missense mutations, commonly present as compound heterozygous. Therefore, specific correction of these point mutations might be a promising therapy for the treatment of PKD patients. We have explored the potential of precise gene editing for the correction of different PKD-causing mutations, using a combination of single-stranded oligodeoxynucleotides (ssODN) with the CRISPR/Cas9 system. We have designed guide RNAs (gRNAs) and single-strand donor templates to target four different PKD-causing mutations in immortalized patient-derived lymphoblastic cell lines, and we have detected the precise correction in three of these mutations. The frequency of the precise gene editing is variable, while the presence of additional insertions/deletions (InDels) has also been detected. Significantly, we have identified high mutation-specificity for two of the PKD-causing mutations. Our results demonstrate the feasibility of a highly personalized gene-editing therapy to treat point mutations in cells derived from PKD patients.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
