Megan L Ratz-Mitchem, Greg Leary, Andrea Grindeland, Derek Silvius, Joseph Guter, Michael P Kavanaugh, Teresa M Gunn
{"title":"痉挛性四肢瘫痪、胼胝体薄和进行性小头畸形(SPATCCM)敲除小鼠模型的生成和表征。","authors":"Megan L Ratz-Mitchem, Greg Leary, Andrea Grindeland, Derek Silvius, Joseph Guter, Michael P Kavanaugh, Teresa M Gunn","doi":"10.1007/s00335-023-10013-4","DOIUrl":null,"url":null,"abstract":"<p><p>Solute carrier family 1 member 4 (SLC1A4), also referred to as Alanine/Serine/Cysteine/Threonine-preferring Transporter 1 (ASCT1), is a sodium-dependent neutral amino acid transporter. It is expressed in many tissues, including the brain, where it is expressed primarily on astrocytes and plays key roles in neuronal differentiation and development, maintaining neurotransmitter homeostasis, and N-methyl-D-aspartate neurotransmission, through regulation of L- and D-serine. Mutations in SLC1A4 are associated with the rare autosomal recessive neurodevelopmental disorder spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM, OMIM 616657). Psychomotor development and speech are significantly impaired in these patients, and many develop seizures. We generated and characterized a knock-in mouse model for the most common mutant allele, which results in a single amino acid change (p.Glu256Lys, or E256K). Homozygous mutants had increased D-serine uptake in the brain, microcephaly, and thin corpus callosum and cortex layer 1. While p.E256K homozygotes showed some significant differences in exploratory behavior relative to wildtype mice, their performance in assays for motor coordination, endurance, learning, and memory was normal, and they showed no significant differences in long-term potentiation. Taken together, these results indicate that the impact of the p.E256K mutation on cognition and motor function is minimal in mice, but other aspects of SLC1A4 function in the brain are conserved. Mice homozygous for p.E256K may be a good model for understanding the developmental basis of the corpus callosum and microcephaly phenotypes observed in SPATCCM patients and assessing whether they are rescued by serine supplementation.</p>","PeriodicalId":18259,"journal":{"name":"Mammalian Genome","volume":" ","pages":"572-585"},"PeriodicalIF":2.7000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10680402/pdf/","citationCount":"0","resultStr":"{\"title\":\"Generation and characterization of a knock-in mouse model for spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM).\",\"authors\":\"Megan L Ratz-Mitchem, Greg Leary, Andrea Grindeland, Derek Silvius, Joseph Guter, Michael P Kavanaugh, Teresa M Gunn\",\"doi\":\"10.1007/s00335-023-10013-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Solute carrier family 1 member 4 (SLC1A4), also referred to as Alanine/Serine/Cysteine/Threonine-preferring Transporter 1 (ASCT1), is a sodium-dependent neutral amino acid transporter. It is expressed in many tissues, including the brain, where it is expressed primarily on astrocytes and plays key roles in neuronal differentiation and development, maintaining neurotransmitter homeostasis, and N-methyl-D-aspartate neurotransmission, through regulation of L- and D-serine. Mutations in SLC1A4 are associated with the rare autosomal recessive neurodevelopmental disorder spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM, OMIM 616657). Psychomotor development and speech are significantly impaired in these patients, and many develop seizures. We generated and characterized a knock-in mouse model for the most common mutant allele, which results in a single amino acid change (p.Glu256Lys, or E256K). Homozygous mutants had increased D-serine uptake in the brain, microcephaly, and thin corpus callosum and cortex layer 1. While p.E256K homozygotes showed some significant differences in exploratory behavior relative to wildtype mice, their performance in assays for motor coordination, endurance, learning, and memory was normal, and they showed no significant differences in long-term potentiation. Taken together, these results indicate that the impact of the p.E256K mutation on cognition and motor function is minimal in mice, but other aspects of SLC1A4 function in the brain are conserved. Mice homozygous for p.E256K may be a good model for understanding the developmental basis of the corpus callosum and microcephaly phenotypes observed in SPATCCM patients and assessing whether they are rescued by serine supplementation.</p>\",\"PeriodicalId\":18259,\"journal\":{\"name\":\"Mammalian Genome\",\"volume\":\" \",\"pages\":\"572-585\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10680402/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Mammalian Genome\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00335-023-10013-4\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/8/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mammalian Genome","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00335-023-10013-4","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/29 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Generation and characterization of a knock-in mouse model for spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM).
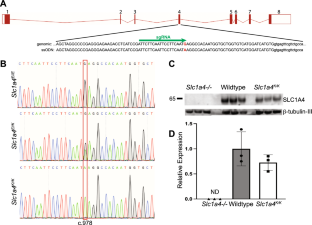
Solute carrier family 1 member 4 (SLC1A4), also referred to as Alanine/Serine/Cysteine/Threonine-preferring Transporter 1 (ASCT1), is a sodium-dependent neutral amino acid transporter. It is expressed in many tissues, including the brain, where it is expressed primarily on astrocytes and plays key roles in neuronal differentiation and development, maintaining neurotransmitter homeostasis, and N-methyl-D-aspartate neurotransmission, through regulation of L- and D-serine. Mutations in SLC1A4 are associated with the rare autosomal recessive neurodevelopmental disorder spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM, OMIM 616657). Psychomotor development and speech are significantly impaired in these patients, and many develop seizures. We generated and characterized a knock-in mouse model for the most common mutant allele, which results in a single amino acid change (p.Glu256Lys, or E256K). Homozygous mutants had increased D-serine uptake in the brain, microcephaly, and thin corpus callosum and cortex layer 1. While p.E256K homozygotes showed some significant differences in exploratory behavior relative to wildtype mice, their performance in assays for motor coordination, endurance, learning, and memory was normal, and they showed no significant differences in long-term potentiation. Taken together, these results indicate that the impact of the p.E256K mutation on cognition and motor function is minimal in mice, but other aspects of SLC1A4 function in the brain are conserved. Mice homozygous for p.E256K may be a good model for understanding the developmental basis of the corpus callosum and microcephaly phenotypes observed in SPATCCM patients and assessing whether they are rescued by serine supplementation.
期刊介绍:
Mammalian Genome focuses on the experimental, theoretical and technical aspects of genetics, genomics, epigenetics and systems biology in mouse, human and other mammalian species, with an emphasis on the relationship between genotype and phenotype, elucidation of biological and disease pathways as well as experimental aspects of interventions, therapeutics, and precision medicine. The journal aims to publish high quality original papers that present novel findings in all areas of mammalian genetic research as well as review articles on areas of topical interest. The journal will also feature commentaries and editorials to inform readers of breakthrough discoveries as well as issues of research standards, policies and ethics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
