Xiangyu Guo, Pernille Sarup, Ahmed Jahoor, Just Jensen, Ole F Christensen
{"title":"代谢组学-基因组学预测可以提高大麦麦芽品质性状的育种价值预测精度。","authors":"Xiangyu Guo, Pernille Sarup, Ahmed Jahoor, Just Jensen, Ole F Christensen","doi":"10.1186/s12711-023-00835-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Metabolomics measures an intermediate stage between genotype and phenotype, and may therefore be useful for breeding. Our objectives were to investigate genetic parameters and accuracies of predicted breeding values for malting quality (MQ) traits when integrating both genomic and metabolomic information. In total, 2430 plots of 562 malting spring barley lines from three years and two locations were included. Five MQ traits were measured in wort produced from each plot. Metabolomic features used were 24,018 nuclear magnetic resonance intensities measured on each wort sample. Methods for statistical analyses were genomic best linear unbiased prediction (GBLUP) and metabolomic-genomic best linear unbiased prediction (MGBLUP). Accuracies of predicted breeding values were compared using two cross-validation strategies: leave-one-year-out (LOYO) and leave-one-line-out (LOLO), and the increase in accuracy from the successive inclusion of first, metabolomic data on the lines in the validation population (VP), and second, both metabolomic data and phenotypes on the lines in the VP, was investigated using the linear regression (LR) method.</p><p><strong>Results: </strong>For all traits, we saw that the metabolome-mediated heritability was substantial. Cross-validation results showed that, in general, prediction accuracies from MGBLUP and GBLUP were similar when phenotypes and metabolomic data were recorded on the same plots. Results from the LR method showed that for all traits, except one, accuracy of MGBLUP increased when including metabolomic data on the lines of the VP, and further increased when including also phenotypes. However, in general the increase in accuracy of MGBLUP when including both metabolomic data and phenotypes on lines of the VP was similar to the increase in accuracy of GBLUP when including phenotypes on the lines of the VP. Therefore, we found that, when metabolomic data were included on the lines of the VP, accuracies substantially increased for lines without phenotypic records, but they did not increase much when phenotypes were already known.</p><p><strong>Conclusions: </strong>MGBLUP is a useful approach to combine phenotypic, genomic and metabolomic data for predicting breeding values for MQ traits. We believe that our results have significant implications for practical breeding of barley and potentially many other species.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"61"},"PeriodicalIF":3.1000,"publicationDate":"2023-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10478459/pdf/","citationCount":"0","resultStr":"{\"title\":\"Metabolomic-genomic prediction can improve prediction accuracy of breeding values for malting quality traits in barley.\",\"authors\":\"Xiangyu Guo, Pernille Sarup, Ahmed Jahoor, Just Jensen, Ole F Christensen\",\"doi\":\"10.1186/s12711-023-00835-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Metabolomics measures an intermediate stage between genotype and phenotype, and may therefore be useful for breeding. Our objectives were to investigate genetic parameters and accuracies of predicted breeding values for malting quality (MQ) traits when integrating both genomic and metabolomic information. In total, 2430 plots of 562 malting spring barley lines from three years and two locations were included. Five MQ traits were measured in wort produced from each plot. Metabolomic features used were 24,018 nuclear magnetic resonance intensities measured on each wort sample. Methods for statistical analyses were genomic best linear unbiased prediction (GBLUP) and metabolomic-genomic best linear unbiased prediction (MGBLUP). Accuracies of predicted breeding values were compared using two cross-validation strategies: leave-one-year-out (LOYO) and leave-one-line-out (LOLO), and the increase in accuracy from the successive inclusion of first, metabolomic data on the lines in the validation population (VP), and second, both metabolomic data and phenotypes on the lines in the VP, was investigated using the linear regression (LR) method.</p><p><strong>Results: </strong>For all traits, we saw that the metabolome-mediated heritability was substantial. Cross-validation results showed that, in general, prediction accuracies from MGBLUP and GBLUP were similar when phenotypes and metabolomic data were recorded on the same plots. Results from the LR method showed that for all traits, except one, accuracy of MGBLUP increased when including metabolomic data on the lines of the VP, and further increased when including also phenotypes. However, in general the increase in accuracy of MGBLUP when including both metabolomic data and phenotypes on lines of the VP was similar to the increase in accuracy of GBLUP when including phenotypes on the lines of the VP. Therefore, we found that, when metabolomic data were included on the lines of the VP, accuracies substantially increased for lines without phenotypic records, but they did not increase much when phenotypes were already known.</p><p><strong>Conclusions: </strong>MGBLUP is a useful approach to combine phenotypic, genomic and metabolomic data for predicting breeding values for MQ traits. We believe that our results have significant implications for practical breeding of barley and potentially many other species.</p>\",\"PeriodicalId\":55120,\"journal\":{\"name\":\"Genetics Selection Evolution\",\"volume\":\"55 1\",\"pages\":\"61\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-09-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10478459/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics Selection Evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12711-023-00835-w\",\"RegionNum\":1,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"AGRICULTURE, DAIRY & ANIMAL SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00835-w","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
Metabolomic-genomic prediction can improve prediction accuracy of breeding values for malting quality traits in barley.
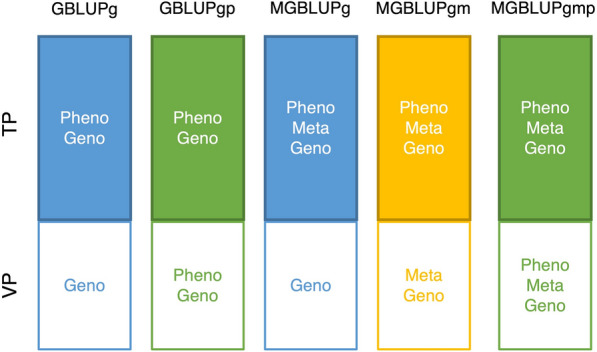
Background: Metabolomics measures an intermediate stage between genotype and phenotype, and may therefore be useful for breeding. Our objectives were to investigate genetic parameters and accuracies of predicted breeding values for malting quality (MQ) traits when integrating both genomic and metabolomic information. In total, 2430 plots of 562 malting spring barley lines from three years and two locations were included. Five MQ traits were measured in wort produced from each plot. Metabolomic features used were 24,018 nuclear magnetic resonance intensities measured on each wort sample. Methods for statistical analyses were genomic best linear unbiased prediction (GBLUP) and metabolomic-genomic best linear unbiased prediction (MGBLUP). Accuracies of predicted breeding values were compared using two cross-validation strategies: leave-one-year-out (LOYO) and leave-one-line-out (LOLO), and the increase in accuracy from the successive inclusion of first, metabolomic data on the lines in the validation population (VP), and second, both metabolomic data and phenotypes on the lines in the VP, was investigated using the linear regression (LR) method.
Results: For all traits, we saw that the metabolome-mediated heritability was substantial. Cross-validation results showed that, in general, prediction accuracies from MGBLUP and GBLUP were similar when phenotypes and metabolomic data were recorded on the same plots. Results from the LR method showed that for all traits, except one, accuracy of MGBLUP increased when including metabolomic data on the lines of the VP, and further increased when including also phenotypes. However, in general the increase in accuracy of MGBLUP when including both metabolomic data and phenotypes on lines of the VP was similar to the increase in accuracy of GBLUP when including phenotypes on the lines of the VP. Therefore, we found that, when metabolomic data were included on the lines of the VP, accuracies substantially increased for lines without phenotypic records, but they did not increase much when phenotypes were already known.
Conclusions: MGBLUP is a useful approach to combine phenotypic, genomic and metabolomic data for predicting breeding values for MQ traits. We believe that our results have significant implications for practical breeding of barley and potentially many other species.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
