Marija J Rowane, Benjamin C Stewart-Bates, Rayna J Doll, Howard J Meyerson, John S Venglarcik, Meghan Callahan, Lauren Fill, Remie Saab, Hans D Ochs, Robert W Hostoffer
{"title":"CD5 b细胞显性原发性免疫缺陷:MAGT1缺陷谱的一部分。","authors":"Marija J Rowane, Benjamin C Stewart-Bates, Rayna J Doll, Howard J Meyerson, John S Venglarcik, Meghan Callahan, Lauren Fill, Remie Saab, Hans D Ochs, Robert W Hostoffer","doi":"10.1177/27534030231199675","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein-Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the \"XMEN\" title pathologies. The updated title, \"X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect,\" was proposed in 2020.</p><p><strong>Objectives: </strong>To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.</p><p><strong>Methods: </strong>Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.</p><p><strong>Design: </strong>Immune re-evaluation done through flow cytometry and next-generation sequencing.</p><p><strong>Results: </strong>Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.</p><p><strong>Conclusion: </strong>We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.</p>","PeriodicalId":75217,"journal":{"name":"Therapeutic advances in allergy and rhinology","volume":"14 ","pages":"27534030231199675"},"PeriodicalIF":0.8000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/c5/10.1177_27534030231199675.PMC10496486.pdf","citationCount":"0","resultStr":"{\"title\":\"CD5 B-Cell Predominant Primary Immunodeficiency: Part of the Spectrum of <i>MAGT1</i> Deficiency.\",\"authors\":\"Marija J Rowane, Benjamin C Stewart-Bates, Rayna J Doll, Howard J Meyerson, John S Venglarcik, Meghan Callahan, Lauren Fill, Remie Saab, Hans D Ochs, Robert W Hostoffer\",\"doi\":\"10.1177/27534030231199675\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein-Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the \\\"XMEN\\\" title pathologies. The updated title, \\\"X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect,\\\" was proposed in 2020.</p><p><strong>Objectives: </strong>To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.</p><p><strong>Methods: </strong>Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.</p><p><strong>Design: </strong>Immune re-evaluation done through flow cytometry and next-generation sequencing.</p><p><strong>Results: </strong>Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.</p><p><strong>Conclusion: </strong>We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.</p>\",\"PeriodicalId\":75217,\"journal\":{\"name\":\"Therapeutic advances in allergy and rhinology\",\"volume\":\"14 \",\"pages\":\"27534030231199675\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/c5/10.1177_27534030231199675.PMC10496486.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Therapeutic advances in allergy and rhinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/27534030231199675\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"0\",\"JCRName\":\"OTORHINOLARYNGOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Therapeutic advances in allergy and rhinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/27534030231199675","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"0","JCRName":"OTORHINOLARYNGOLOGY","Score":null,"Total":0}
CD5 B-Cell Predominant Primary Immunodeficiency: Part of the Spectrum of MAGT1 Deficiency.
Background: Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein-Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the "XMEN" title pathologies. The updated title, "X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect," was proposed in 2020.
Objectives: To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.
Methods: Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.
Design: Immune re-evaluation done through flow cytometry and next-generation sequencing.
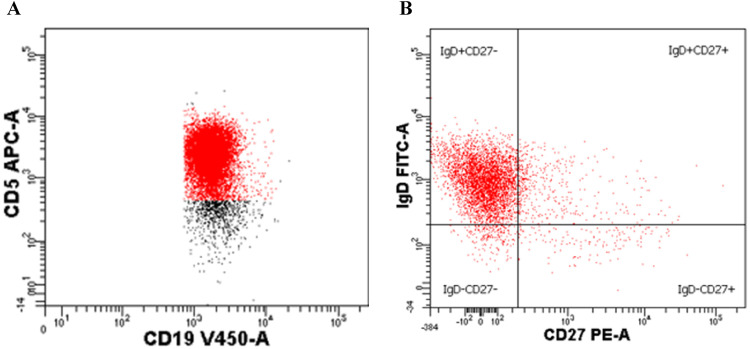
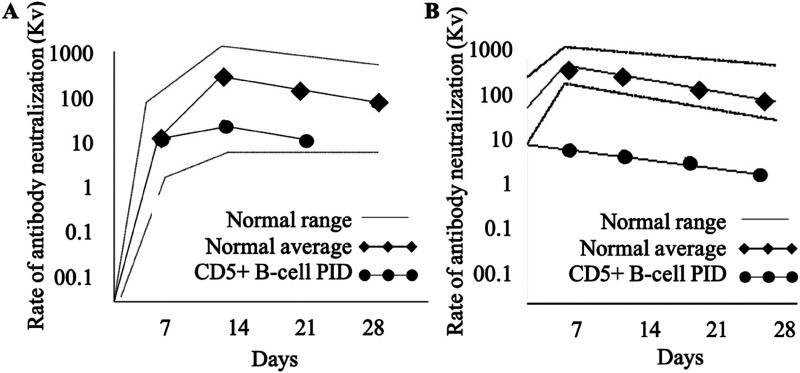
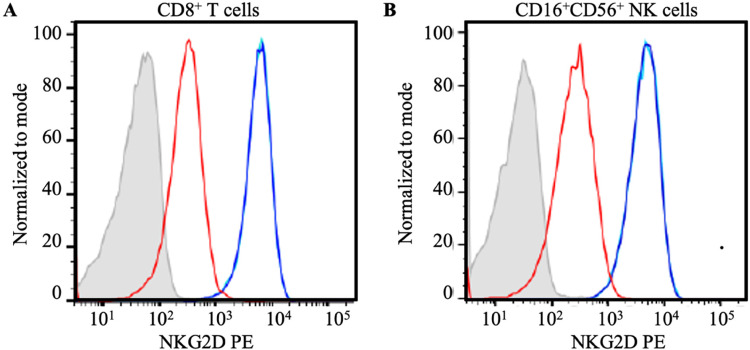
Results: Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.
Conclusion: We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
