Friederike T Füsser, Jan Wollenhaupt, Manfred S Weiss, Daniel Kümmel, Oliver Koch
{"title":"利用结晶学片段筛选确定的针对分枝杆菌硫氧还蛋白还原酶的基于片段的药物设计的新起点。","authors":"Friederike T Füsser, Jan Wollenhaupt, Manfred S Weiss, Daniel Kümmel, Oliver Koch","doi":"10.1107/S2059798323005223","DOIUrl":null,"url":null,"abstract":"<p><p>The increasing number of people dying from tuberculosis and the existence of extensively drug-resistant strains has led to an urgent need for new antituberculotic drugs with alternative modes of action. As part of the thioredoxin system, thioredoxin reductase (TrxR) is essential for the survival of Mycobacterium tuberculosis (Mtb) and shows substantial differences from human TrxR, making it a promising and most likely selective target. As a model organism for Mtb, crystals of Mycobacterium smegmatis TrxR that diffracted to high resolution were used in crystallographic fragment screening to discover binding fragments and new binding sites. The application of the 96 structurally diverse fragments from the F2X-Entry Screen revealed 56 new starting points for fragment-based drug design of new TrxR inhibitors. Over 200 crystal structures were analyzed using FragMAXapp, which includes processing and refinement by largely automated software pipelines and hit identification via the multi-data-set analysis approach PanDDA. The fragments are bound to 11 binding sites, of which four are positioned at binding pockets or important interaction sites and therefore show high potential for possible inhibition of TrxR.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":"79 Pt 9","pages":"857-865"},"PeriodicalIF":2.6000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10478635/pdf/","citationCount":"0","resultStr":"{\"title\":\"Novel starting points for fragment-based drug design against mycobacterial thioredoxin reductase identified using crystallographic fragment screening.\",\"authors\":\"Friederike T Füsser, Jan Wollenhaupt, Manfred S Weiss, Daniel Kümmel, Oliver Koch\",\"doi\":\"10.1107/S2059798323005223\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The increasing number of people dying from tuberculosis and the existence of extensively drug-resistant strains has led to an urgent need for new antituberculotic drugs with alternative modes of action. As part of the thioredoxin system, thioredoxin reductase (TrxR) is essential for the survival of Mycobacterium tuberculosis (Mtb) and shows substantial differences from human TrxR, making it a promising and most likely selective target. As a model organism for Mtb, crystals of Mycobacterium smegmatis TrxR that diffracted to high resolution were used in crystallographic fragment screening to discover binding fragments and new binding sites. The application of the 96 structurally diverse fragments from the F2X-Entry Screen revealed 56 new starting points for fragment-based drug design of new TrxR inhibitors. Over 200 crystal structures were analyzed using FragMAXapp, which includes processing and refinement by largely automated software pipelines and hit identification via the multi-data-set analysis approach PanDDA. The fragments are bound to 11 binding sites, of which four are positioned at binding pockets or important interaction sites and therefore show high potential for possible inhibition of TrxR.</p>\",\"PeriodicalId\":7116,\"journal\":{\"name\":\"Acta Crystallographica. Section D, Structural Biology\",\"volume\":\"79 Pt 9\",\"pages\":\"857-865\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2023-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10478635/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Crystallographica. Section D, Structural Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1107/S2059798323005223\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798323005223","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Novel starting points for fragment-based drug design against mycobacterial thioredoxin reductase identified using crystallographic fragment screening.
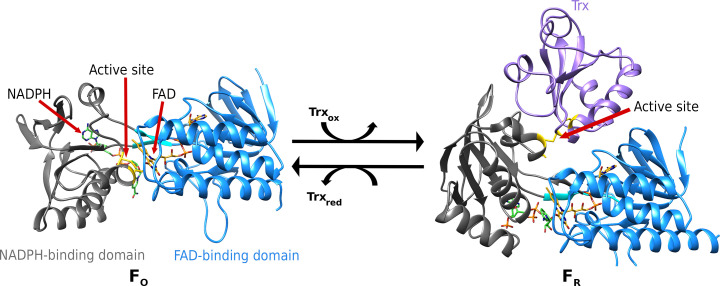
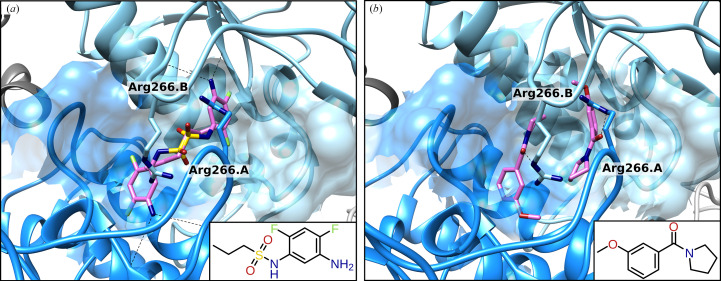
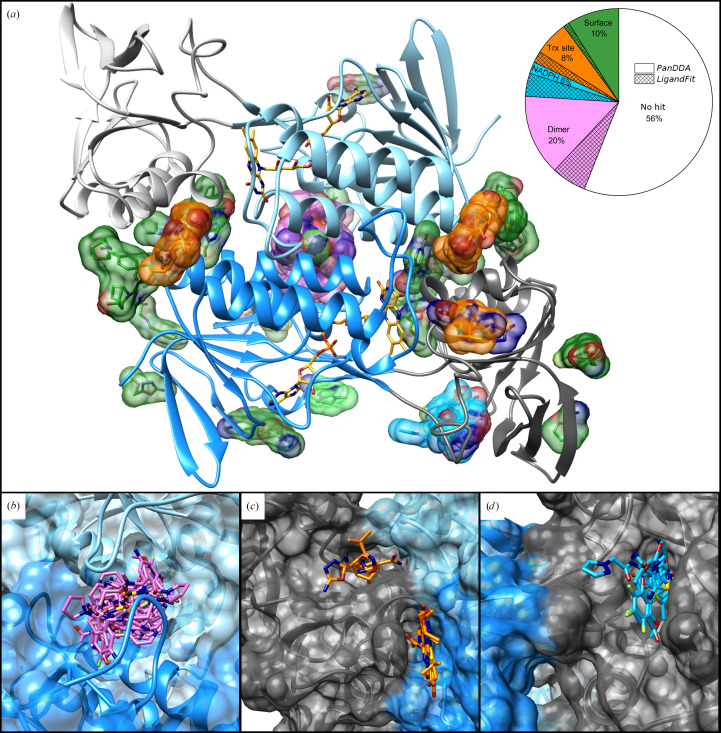
The increasing number of people dying from tuberculosis and the existence of extensively drug-resistant strains has led to an urgent need for new antituberculotic drugs with alternative modes of action. As part of the thioredoxin system, thioredoxin reductase (TrxR) is essential for the survival of Mycobacterium tuberculosis (Mtb) and shows substantial differences from human TrxR, making it a promising and most likely selective target. As a model organism for Mtb, crystals of Mycobacterium smegmatis TrxR that diffracted to high resolution were used in crystallographic fragment screening to discover binding fragments and new binding sites. The application of the 96 structurally diverse fragments from the F2X-Entry Screen revealed 56 new starting points for fragment-based drug design of new TrxR inhibitors. Over 200 crystal structures were analyzed using FragMAXapp, which includes processing and refinement by largely automated software pipelines and hit identification via the multi-data-set analysis approach PanDDA. The fragments are bound to 11 binding sites, of which four are positioned at binding pockets or important interaction sites and therefore show high potential for possible inhibition of TrxR.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
