{"title":"肺腺癌淋巴结转移相关预后特征的建立。","authors":"Jiao Yu, Gang Li, Yingxuan Tian, Shufen Huo","doi":"10.1155/2023/6585109","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lung adenocarcinoma (LUAD) is the most common histological subtype of non-small cell lung cancer (NSCLC) with a low 5-year survival rate, which may be associated with the presence of metastatic tumors at the time of diagnosis, especially lymph node metastasis (LNM). This study aimed to construct a LNM-related gene signature for predicting the prognosis of patients with LUAD.</p><p><strong>Methods: </strong>RNA sequencing data and clinical information of LUAD patients were extracted from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Samples were divided into metastasis (M) and nonmetastasis (NM) groups based on LNM status. Differentially expressed genes (DEGs) between M and NM groups were screened, and then WGCNA was applied to identify key genes. Furthermore, univariate Cox and LASSO regression analyses were conducted to construct a risk score model, and the predictive performance of model was validated by GSE68465, GSE42127, and GSE50081. The protein and mRNA expression level of LNM-associated genes were detected by human protein atlas (HPA) and GSE68465.</p><p><strong>Results: </strong>A prognostic model based on eight LNM-related genes (ANGPTL4, BARX2, GPR98, KRT6A, PTPRH, RGS20, TCN1, and TNS4) was developed. Patients in the high-risk group had poorer overall survival than those in the low-risk group, and validation analysis showed that this model had potential predictive value for patients with LUAD. HPA analysis supported the upregulation of ANGPTL4, KRT6A, BARX2, RGS20 and the downregulation of GPR98 in LUAD compared with normal tissues.</p><p><strong>Conclusion: </strong>Our results indicated that the eight LNM-related genes signature had potential value in the prognosis of patients with LUAD, which may have important practical implications.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2023 ","pages":"6585109"},"PeriodicalIF":2.1000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9904923/pdf/","citationCount":"3","resultStr":"{\"title\":\"Establishment of a Lymph Node Metastasis-Associated Prognostic Signature for Lung Adenocarcinoma.\",\"authors\":\"Jiao Yu, Gang Li, Yingxuan Tian, Shufen Huo\",\"doi\":\"10.1155/2023/6585109\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Lung adenocarcinoma (LUAD) is the most common histological subtype of non-small cell lung cancer (NSCLC) with a low 5-year survival rate, which may be associated with the presence of metastatic tumors at the time of diagnosis, especially lymph node metastasis (LNM). This study aimed to construct a LNM-related gene signature for predicting the prognosis of patients with LUAD.</p><p><strong>Methods: </strong>RNA sequencing data and clinical information of LUAD patients were extracted from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Samples were divided into metastasis (M) and nonmetastasis (NM) groups based on LNM status. Differentially expressed genes (DEGs) between M and NM groups were screened, and then WGCNA was applied to identify key genes. Furthermore, univariate Cox and LASSO regression analyses were conducted to construct a risk score model, and the predictive performance of model was validated by GSE68465, GSE42127, and GSE50081. The protein and mRNA expression level of LNM-associated genes were detected by human protein atlas (HPA) and GSE68465.</p><p><strong>Results: </strong>A prognostic model based on eight LNM-related genes (ANGPTL4, BARX2, GPR98, KRT6A, PTPRH, RGS20, TCN1, and TNS4) was developed. Patients in the high-risk group had poorer overall survival than those in the low-risk group, and validation analysis showed that this model had potential predictive value for patients with LUAD. HPA analysis supported the upregulation of ANGPTL4, KRT6A, BARX2, RGS20 and the downregulation of GPR98 in LUAD compared with normal tissues.</p><p><strong>Conclusion: </strong>Our results indicated that the eight LNM-related genes signature had potential value in the prognosis of patients with LUAD, which may have important practical implications.</p>\",\"PeriodicalId\":12778,\"journal\":{\"name\":\"Genetics research\",\"volume\":\"2023 \",\"pages\":\"6585109\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9904923/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/6585109\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/2023/6585109","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 3
摘要
背景:肺腺癌(LUAD)是非小细胞肺癌(NSCLC)中最常见的组织学亚型,其5年生存率较低,可能与诊断时存在转移性肿瘤,尤其是淋巴结转移(LNM)有关。本研究旨在构建预测LUAD患者预后的lnm相关基因标记。方法:从Cancer Genome Atlas (TCGA)和Gene Expression Omnibus (GEO)数据库中提取LUAD患者的RNA测序数据和临床信息。根据肿瘤转移状态将样本分为转移(M)组和非转移(NM)组。筛选M组和NM组之间的差异表达基因(DEGs),然后应用WGCNA鉴定关键基因。通过单变量Cox和LASSO回归分析构建风险评分模型,并通过GSE68465、GSE42127和GSE50081对模型的预测性能进行验证。采用人蛋白图谱(human protein atlas, HPA)和GSE68465检测lnm相关基因的蛋白表达和mRNA表达水平。结果:建立了基于8个lnm相关基因(ANGPTL4、BARX2、GPR98、KRT6A、PTPRH、RGS20、TCN1和TNS4)的预后模型。高危组患者的总生存率低于低危组,验证分析表明该模型对LUAD患者具有潜在的预测价值。HPA分析支持LUAD与正常组织相比ANGPTL4、KRT6A、BARX2、RGS20表达上调,GPR98表达下调。结论:我们的研究结果表明,8个lnm相关基因标记对LUAD患者的预后有潜在价值,可能具有重要的现实意义。
Establishment of a Lymph Node Metastasis-Associated Prognostic Signature for Lung Adenocarcinoma.
Background: Lung adenocarcinoma (LUAD) is the most common histological subtype of non-small cell lung cancer (NSCLC) with a low 5-year survival rate, which may be associated with the presence of metastatic tumors at the time of diagnosis, especially lymph node metastasis (LNM). This study aimed to construct a LNM-related gene signature for predicting the prognosis of patients with LUAD.
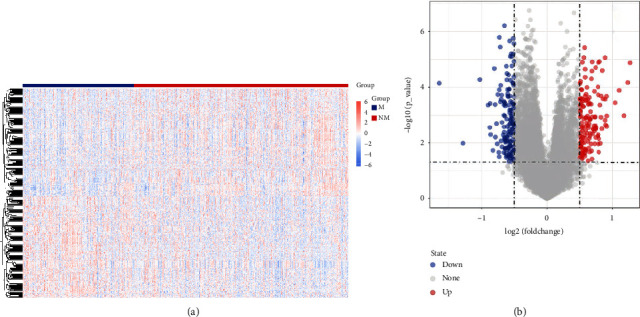
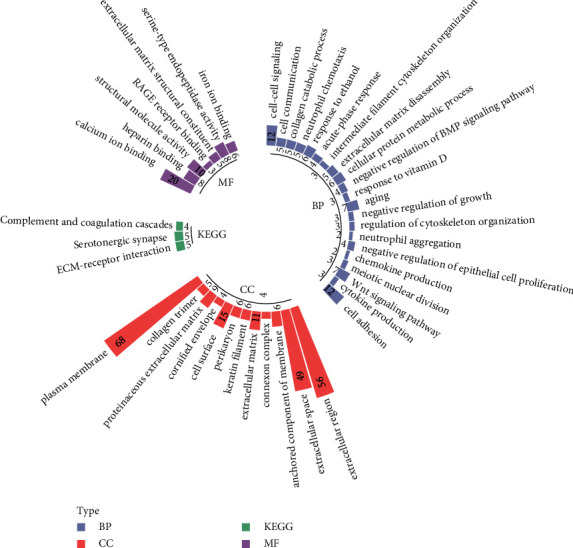
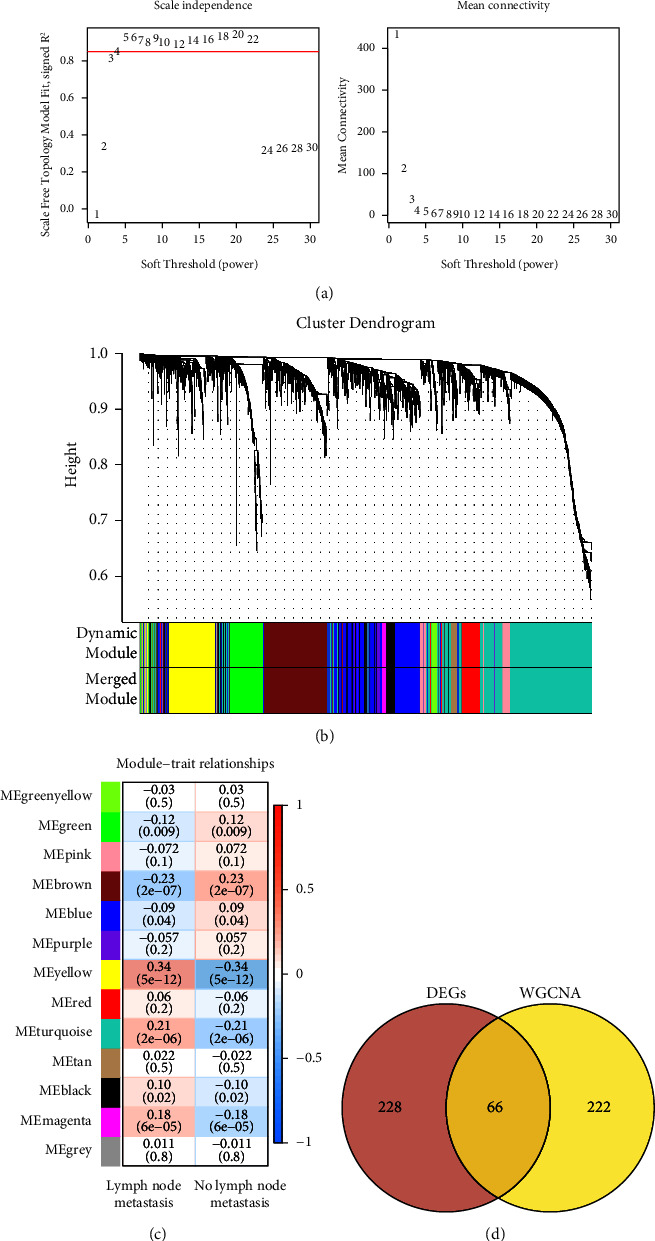
Methods: RNA sequencing data and clinical information of LUAD patients were extracted from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Samples were divided into metastasis (M) and nonmetastasis (NM) groups based on LNM status. Differentially expressed genes (DEGs) between M and NM groups were screened, and then WGCNA was applied to identify key genes. Furthermore, univariate Cox and LASSO regression analyses were conducted to construct a risk score model, and the predictive performance of model was validated by GSE68465, GSE42127, and GSE50081. The protein and mRNA expression level of LNM-associated genes were detected by human protein atlas (HPA) and GSE68465.
Results: A prognostic model based on eight LNM-related genes (ANGPTL4, BARX2, GPR98, KRT6A, PTPRH, RGS20, TCN1, and TNS4) was developed. Patients in the high-risk group had poorer overall survival than those in the low-risk group, and validation analysis showed that this model had potential predictive value for patients with LUAD. HPA analysis supported the upregulation of ANGPTL4, KRT6A, BARX2, RGS20 and the downregulation of GPR98 in LUAD compared with normal tissues.
Conclusion: Our results indicated that the eight LNM-related genes signature had potential value in the prognosis of patients with LUAD, which may have important practical implications.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
