Janne Austestad, Tor Magne Madland, Miriam Sandnes, Torjan Magne Haslerud, Andreas Benneche, Håkon Reikvam
{"title":"骨髓增生性肿瘤患者的 VEXAS 综合征。","authors":"Janne Austestad, Tor Magne Madland, Miriam Sandnes, Torjan Magne Haslerud, Andreas Benneche, Håkon Reikvam","doi":"10.1155/2023/6551544","DOIUrl":null,"url":null,"abstract":"<p><p>VEXAS syndrome stands for vacuoles, E1 enzyme, <i>X</i>-linked, autoinflammatory, somatic syndrome. The syndrome is a combined hematological and rheumatological condition caused by a somatic mutation in the <i>UBA1</i>. There is an association between VEXAS and hematological conditions such as myelodysplastic syndrome (MDS), monoclonal gammopathies of uncertain conditions (MGUS), multiple myeloma (MM), and monoclonal B-cell lymphoproliferative conditions. There are not many descriptions of patients having VEXAS in combination with myeloproliferative neoplasm (MPN). With this article, we want to present a case history of a man in his sixties with a <i>JAK2</i>V617F mutated essential thrombocythemia (ET) developing VEXAS syndrome. The inflammatory symptoms occurred three and a half years after the ET diagnosis. He started to experience symptoms of autoinflammation and an overall worsening of his health, and blood work showed high inflammatory markers, leading to repeated hospitalizations. His major complaint was stiffness and pain, and high dosages of prednisolone were necessary to obtain pain relief. He subsequently developed anemia and significantly variable levels of thrombocytes, which previously were at a steady level. To evaluate his ET, we made a bone marrow smear demonstrating vacuolated myeloid and erythroid cells. Having VEXAS syndrome in mind, genetic testing identifying the <i>UBA1</i> gene mutation was performed, thus confirming our suspicion. The work-up with myeloid panel on his bone marrow identified genetic mutation in the <i>DNMT3</i> too. After developing VEXAS syndrome, he experienced thromboembolic events with both cerebral infarction and pulmonary embolism. Thromboembolic events are also common in <i>JAK2</i> mutated patients, but in his case, they presented first after VEXAS had developed. Throughout the course of his condition, several attempts with prednisolone tapering and steroid sparing drugs were tried. He could not get pain relief unless the combination of medications included a relatively high dose of prednisolone. Currently, the patient uses prednisolone, anagrelide, and ruxolitinib, with partial remission and fewer hospitalizations and more stabilized hemoglobin and thrombocytes.</p>","PeriodicalId":46307,"journal":{"name":"Case Reports in Hematology","volume":"2023 ","pages":"6551544"},"PeriodicalIF":0.7000,"publicationDate":"2023-02-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9985496/pdf/","citationCount":"0","resultStr":"{\"title\":\"VEXAS Syndrome in a Patient with Myeloproliferative Neoplasia.\",\"authors\":\"Janne Austestad, Tor Magne Madland, Miriam Sandnes, Torjan Magne Haslerud, Andreas Benneche, Håkon Reikvam\",\"doi\":\"10.1155/2023/6551544\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>VEXAS syndrome stands for vacuoles, E1 enzyme, <i>X</i>-linked, autoinflammatory, somatic syndrome. The syndrome is a combined hematological and rheumatological condition caused by a somatic mutation in the <i>UBA1</i>. There is an association between VEXAS and hematological conditions such as myelodysplastic syndrome (MDS), monoclonal gammopathies of uncertain conditions (MGUS), multiple myeloma (MM), and monoclonal B-cell lymphoproliferative conditions. There are not many descriptions of patients having VEXAS in combination with myeloproliferative neoplasm (MPN). With this article, we want to present a case history of a man in his sixties with a <i>JAK2</i>V617F mutated essential thrombocythemia (ET) developing VEXAS syndrome. The inflammatory symptoms occurred three and a half years after the ET diagnosis. He started to experience symptoms of autoinflammation and an overall worsening of his health, and blood work showed high inflammatory markers, leading to repeated hospitalizations. His major complaint was stiffness and pain, and high dosages of prednisolone were necessary to obtain pain relief. He subsequently developed anemia and significantly variable levels of thrombocytes, which previously were at a steady level. To evaluate his ET, we made a bone marrow smear demonstrating vacuolated myeloid and erythroid cells. Having VEXAS syndrome in mind, genetic testing identifying the <i>UBA1</i> gene mutation was performed, thus confirming our suspicion. The work-up with myeloid panel on his bone marrow identified genetic mutation in the <i>DNMT3</i> too. After developing VEXAS syndrome, he experienced thromboembolic events with both cerebral infarction and pulmonary embolism. Thromboembolic events are also common in <i>JAK2</i> mutated patients, but in his case, they presented first after VEXAS had developed. Throughout the course of his condition, several attempts with prednisolone tapering and steroid sparing drugs were tried. He could not get pain relief unless the combination of medications included a relatively high dose of prednisolone. Currently, the patient uses prednisolone, anagrelide, and ruxolitinib, with partial remission and fewer hospitalizations and more stabilized hemoglobin and thrombocytes.</p>\",\"PeriodicalId\":46307,\"journal\":{\"name\":\"Case Reports in Hematology\",\"volume\":\"2023 \",\"pages\":\"6551544\"},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2023-02-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9985496/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Hematology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/6551544\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Hematology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/6551544","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
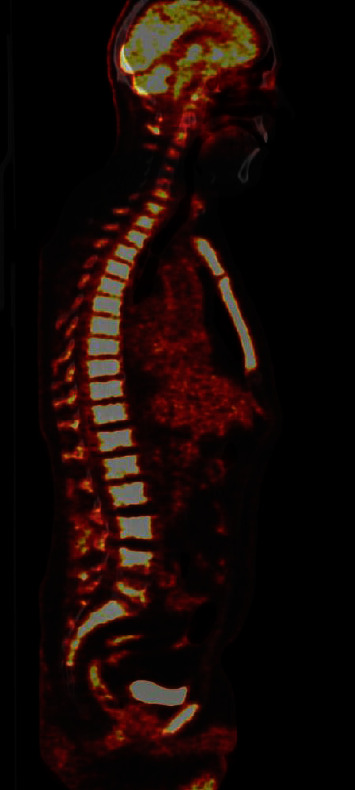
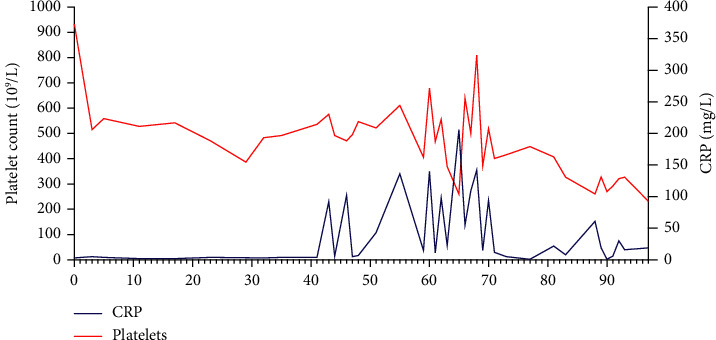
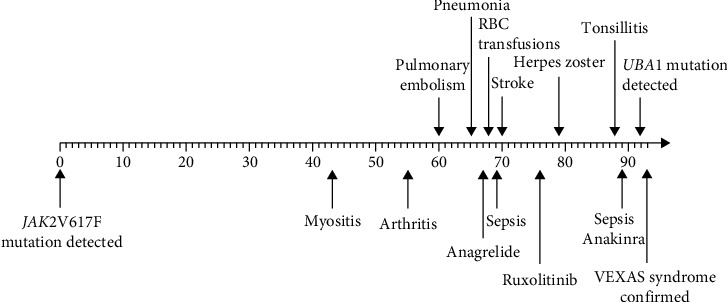
VEXAS 综合征是空泡、E1 酶、X 连锁、自身炎症、体细胞综合征的缩写。该综合征是一种由 UBA1 体细胞突变引起的血液病和风湿病综合症。VEXAS 与骨髓增生异常综合征(MDS)、病情不确定的单克隆丙种球蛋白病(MGUS)、多发性骨髓瘤(MM)和单克隆 B 细胞淋巴增生性疾病等血液病有关联。关于VEXAS合并骨髓增生性肿瘤(MPN)患者的描述并不多。通过这篇文章,我们想介绍一例60多岁男性患者的病史,他患有JAK2V617F突变的原发性血小板增多症(ET),并发展为VEXAS综合征。炎症症状发生在 ET 诊断三年半之后。他开始出现自身炎症症状,健康状况全面恶化,血液检查显示炎症标志物偏高,导致多次住院治疗。他的主要症状是身体僵硬和疼痛,必须使用大剂量泼尼松龙才能缓解疼痛。随后,他出现了贫血,血小板水平也明显变化,而之前血小板水平一直很稳定。为了评估他的 ET,我们做了骨髓涂片,结果显示骨髓细胞和红细胞空泡化。考虑到 VEXAS 综合征,我们进行了基因检测,发现 UBA1 基因突变,从而证实了我们的怀疑。在对他的骨髓进行髓系检查时,也发现了 DNMT3 基因突变。在患上 VEXAS 综合征后,他经历了血栓栓塞事件,包括脑梗塞和肺栓塞。血栓栓塞事件在JAK2基因突变的患者中也很常见,但在他的病例中,血栓栓塞事件是在VEXAS综合征发生后首先出现的。在他的整个病程中,曾多次尝试减少泼尼松龙用量和使用类固醇类药物。除非药物组合中包括相对高剂量的泼尼松龙,否则他的疼痛无法缓解。目前,患者使用泼尼松龙、阿那格雷和鲁索利替尼,病情得到部分缓解,住院次数减少,血红蛋白和血小板也更加稳定。
VEXAS Syndrome in a Patient with Myeloproliferative Neoplasia.
VEXAS syndrome stands for vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome. The syndrome is a combined hematological and rheumatological condition caused by a somatic mutation in the UBA1. There is an association between VEXAS and hematological conditions such as myelodysplastic syndrome (MDS), monoclonal gammopathies of uncertain conditions (MGUS), multiple myeloma (MM), and monoclonal B-cell lymphoproliferative conditions. There are not many descriptions of patients having VEXAS in combination with myeloproliferative neoplasm (MPN). With this article, we want to present a case history of a man in his sixties with a JAK2V617F mutated essential thrombocythemia (ET) developing VEXAS syndrome. The inflammatory symptoms occurred three and a half years after the ET diagnosis. He started to experience symptoms of autoinflammation and an overall worsening of his health, and blood work showed high inflammatory markers, leading to repeated hospitalizations. His major complaint was stiffness and pain, and high dosages of prednisolone were necessary to obtain pain relief. He subsequently developed anemia and significantly variable levels of thrombocytes, which previously were at a steady level. To evaluate his ET, we made a bone marrow smear demonstrating vacuolated myeloid and erythroid cells. Having VEXAS syndrome in mind, genetic testing identifying the UBA1 gene mutation was performed, thus confirming our suspicion. The work-up with myeloid panel on his bone marrow identified genetic mutation in the DNMT3 too. After developing VEXAS syndrome, he experienced thromboembolic events with both cerebral infarction and pulmonary embolism. Thromboembolic events are also common in JAK2 mutated patients, but in his case, they presented first after VEXAS had developed. Throughout the course of his condition, several attempts with prednisolone tapering and steroid sparing drugs were tried. He could not get pain relief unless the combination of medications included a relatively high dose of prednisolone. Currently, the patient uses prednisolone, anagrelide, and ruxolitinib, with partial remission and fewer hospitalizations and more stabilized hemoglobin and thrombocytes.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
