{"title":"从实时时变密度泛函理论耦合到Ehrenfest方案的振动动力学:以对香豆酸为例","authors":"Michele Guerrini, Jannis Krumland, Caterina Cocchi","doi":"10.1007/s00214-023-03036-2","DOIUrl":null,"url":null,"abstract":"Abstract We investigate the vibronic dynamics of a modified version of the p-coumaric acid using real-time time-dependent density-functional theory coupled with the Ehrenfest scheme in the adiabatic local density approximation. Due to the issues of this functional to yield a reliable starting point for the evolution of the electron-nuclear system triggered by a pulse, we start off the simulations constraining the electronic occupation of the molecule in two excited states corresponding to a bright, delocalized transition, and a dark, charge-transfer-like excitation. By monitoring the kinetic energy spectral density, we analyze the nature of the nuclear motion over a time window of 300 fs. Anharmonic effects appear at low frequencies, below 500 cm $$^{-1}$$ <mml:math xmlns:mml=\"http://www.w3.org/1998/Math/MathML\"> <mml:msup> <mml:mrow /> <mml:mrow> <mml:mo>-</mml:mo> <mml:mn>1</mml:mn> </mml:mrow> </mml:msup> </mml:math> , and are particularly pronounced in the charge-transfer excitation. In this case, after about 200 fs, the molecular backbone becomes largely distorted and the initially constrained occupations evolve toward a different electronic configuration. On the other hand, the dynamics initialized from the delocalized bright excitation are electronically and structurally stable, and the resulting nuclear motion is markedly harmonic. Our results provide indications to decipher the vibronic dynamics of this chromophore and related systems in view of more elaborated simulations embedding the molecules in a realistic environment.","PeriodicalId":23045,"journal":{"name":"Theoretical Chemistry Accounts","volume":"7 1","pages":"0"},"PeriodicalIF":1.8000,"publicationDate":"2023-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Vibronic dynamics from real-time time-dependent density-functional theory coupled to the Ehrenfest scheme: the example of p-coumaric acid\",\"authors\":\"Michele Guerrini, Jannis Krumland, Caterina Cocchi\",\"doi\":\"10.1007/s00214-023-03036-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Abstract We investigate the vibronic dynamics of a modified version of the p-coumaric acid using real-time time-dependent density-functional theory coupled with the Ehrenfest scheme in the adiabatic local density approximation. Due to the issues of this functional to yield a reliable starting point for the evolution of the electron-nuclear system triggered by a pulse, we start off the simulations constraining the electronic occupation of the molecule in two excited states corresponding to a bright, delocalized transition, and a dark, charge-transfer-like excitation. By monitoring the kinetic energy spectral density, we analyze the nature of the nuclear motion over a time window of 300 fs. Anharmonic effects appear at low frequencies, below 500 cm $$^{-1}$$ <mml:math xmlns:mml=\\\"http://www.w3.org/1998/Math/MathML\\\"> <mml:msup> <mml:mrow /> <mml:mrow> <mml:mo>-</mml:mo> <mml:mn>1</mml:mn> </mml:mrow> </mml:msup> </mml:math> , and are particularly pronounced in the charge-transfer excitation. In this case, after about 200 fs, the molecular backbone becomes largely distorted and the initially constrained occupations evolve toward a different electronic configuration. On the other hand, the dynamics initialized from the delocalized bright excitation are electronically and structurally stable, and the resulting nuclear motion is markedly harmonic. Our results provide indications to decipher the vibronic dynamics of this chromophore and related systems in view of more elaborated simulations embedding the molecules in a realistic environment.\",\"PeriodicalId\":23045,\"journal\":{\"name\":\"Theoretical Chemistry Accounts\",\"volume\":\"7 1\",\"pages\":\"0\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2023-10-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Theoretical Chemistry Accounts\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s00214-023-03036-2\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Chemistry Accounts","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s00214-023-03036-2","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Vibronic dynamics from real-time time-dependent density-functional theory coupled to the Ehrenfest scheme: the example of p-coumaric acid
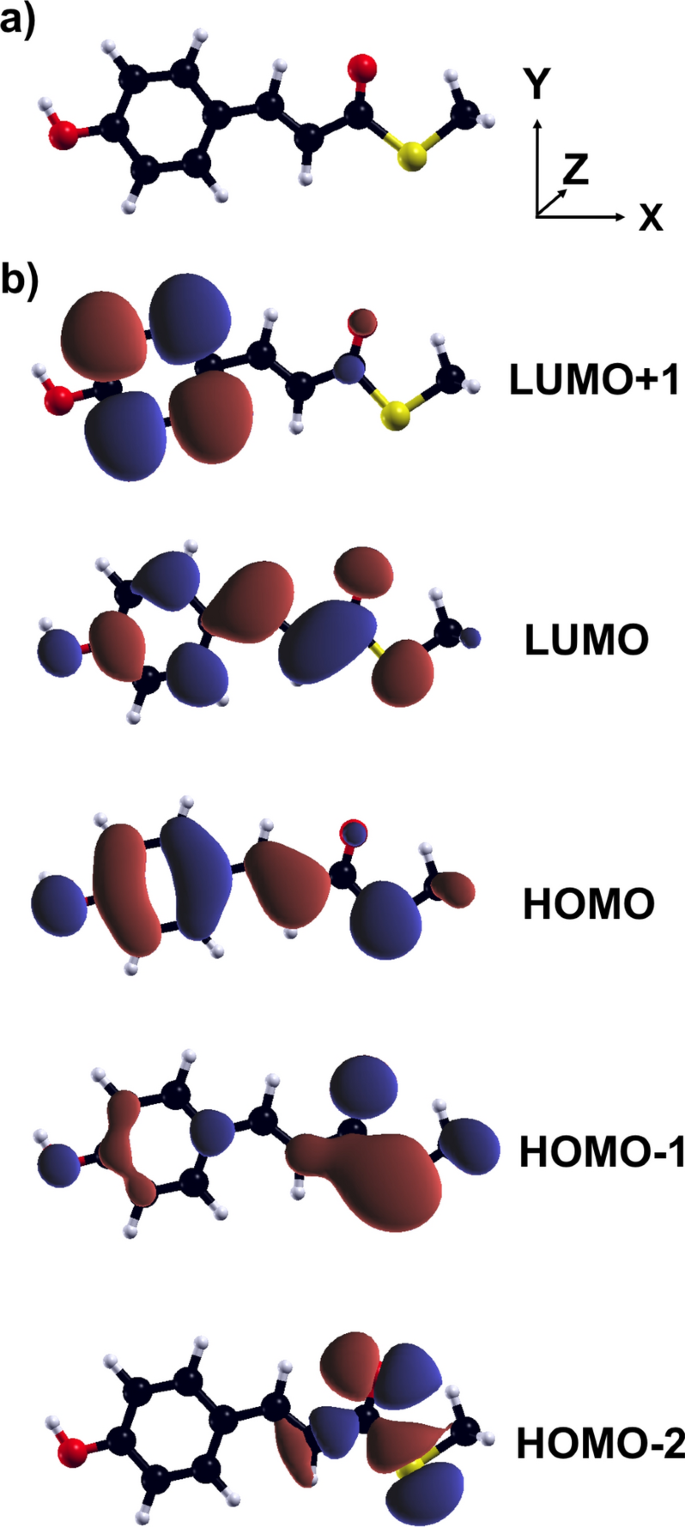
Abstract We investigate the vibronic dynamics of a modified version of the p-coumaric acid using real-time time-dependent density-functional theory coupled with the Ehrenfest scheme in the adiabatic local density approximation. Due to the issues of this functional to yield a reliable starting point for the evolution of the electron-nuclear system triggered by a pulse, we start off the simulations constraining the electronic occupation of the molecule in two excited states corresponding to a bright, delocalized transition, and a dark, charge-transfer-like excitation. By monitoring the kinetic energy spectral density, we analyze the nature of the nuclear motion over a time window of 300 fs. Anharmonic effects appear at low frequencies, below 500 cm $$^{-1}$$ -1 , and are particularly pronounced in the charge-transfer excitation. In this case, after about 200 fs, the molecular backbone becomes largely distorted and the initially constrained occupations evolve toward a different electronic configuration. On the other hand, the dynamics initialized from the delocalized bright excitation are electronically and structurally stable, and the resulting nuclear motion is markedly harmonic. Our results provide indications to decipher the vibronic dynamics of this chromophore and related systems in view of more elaborated simulations embedding the molecules in a realistic environment.
期刊介绍:
TCA publishes papers in all fields of theoretical chemistry, computational chemistry, and modeling. Fundamental studies as well as applications are included in the scope. In many cases, theorists and computational chemists have special concerns which reach either across the vertical borders of the special disciplines in chemistry or else across the horizontal borders of structure, spectra, synthesis, and dynamics. TCA is especially interested in papers that impact upon multiple chemical disciplines.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
